
The potato cyst nematodes (PCNs), Globodera pallida (pale cyst nematode) and G. rostochiensis (golden cyst nematode), are sedentary endoparasites of solanaceous crops and among the most economically important nematode pests of potato worldwide. G. pallida and G. rostochiensis are well-recognized quarantine pests capable of causing yield losses up to 80% in susceptible potato cultivars (Contina et al., 2019; Talavera et al., 1998). Their long-term survival as cysts in soil, combined with ease of spread through infested soil, seed tubers, and farm equipment, makes their control challenging (Jones et al., 2013; Price et al., 2021). In the United States, G. rostochiensis is established in New York, whereas G. pallida is currently confined to Idaho, and both species are under intensive containment and eradication programs by USDA-APHIS (USDA-APHIS, 2009a; USDA-APHIS, 2009b).
In 2008, Globodera ellingtonae was described from Oregon and Idaho and was later detected in South America (Handoo et al., 2012; Subbotin et al., 2020). Initial studies revealed a close phylogenetic relationship between this species and other PCNs (G. pallida and G. rostochiensis) and demonstrated that G. ellingtonae can reproduce on potato; however, it has not yet been reported to cause significant economic losses and may occur in mixed populations (APHIS-USDA, 2009; Handoo et al., 2012; National Agricultural Statistics Service, 2023; Zasada et al., 2019). Such mixed infestations complicate species identification and raise concerns about regulatory misclassification, particularly when diagnostic outcomes drive quarantine decisions and management actions.
Differentiating the Globodera species solely on morphology is difficult because of overlapping characters among eggs, second-stage juveniles, and cysts. Subtle morphological differences, intraspecific variability, and the frequent co-occurrence of species all contribute to misidentification (Subbotin et al., 2001; Wainer and Dinh, 2021). In addition to morphological identification, molecular diagnostics may provide confirmation for morphological identification and be a useful and reliable tool for species identification. So far, molecular tools for Globodera spp. include conventional polymerase chain reaction (PCR) with species-specific primers, restriction fragment length polymorphism-PCR (RFLP-PCR), multiplex PCR, and sequencing of rDNA and mitochondrial markers, and, more recently, quantitative PCR (qPCR)-based assays (Madani et al., 2008; Nakhla et al., 2010; Xu et al., 2023). For G. ellingtonae, a limited number of assays based on ribosomal and nuclear loci (including qPCR/TaqMan probes) have been developed and used in regulatory and survey contexts; however, these methods rely on specialized thermocyclers, and real-time PCR platforms and typically require a relatively high number of cysts for reliable DNA extraction (Chronis et al., 2014; Lax et al., 2014).
Loop-mediated isothermal amplification (LAMP), first introduced in 2000, is a rapid and sensitive molecular technique that enables DNA amplification under constant isothermal conditions (Ahuja et al., 2021; Babilônia et al., 2024). The method utilizes strand displacement activity with four to six primers designed to recognize six to eight distinct regions within the target DNA. LAMP reactions produce multiple amplicons in 15 min to 1 h, offering a fast and effective alternative to conventional PCR-based diagnostics (Ahuja and Somvanshi, 2021; Babilônia et al., 2024; Singh et al., 2021). Detection of LAMP products can be achieved through various methods, fluorescence-based indicators (SYBR safe, fluorescent detection reagent (FDR), SYBR Green I) (Ezzatyhusna et al., 2017; Thita et al., 2019; Yang et al., 2012), turbidity-based precipitation (through magnesium pyrophosphate precipitation), using colorimetric dyes (such as hydroxy naphthol blue, malachite green, and SYBR Green I) (Sriworarat et al., 2015; Sukphattanaudomchoke et al., 2020), and nanoparticle-based precipitation (Ruang-areerate et al., 2021; Suwannin et al., 2021). Due to its simplicity and versatility, LAMP assay has been widely adopted in different fields including plant pathology, microbiology, virology, medicine, and forensics sciences (Babilônia et al., 2024; Mhatre et al., 2022; Vieira and Gleason, 2019).
The first LAMP assay developed for plant parasitic nematodes was for Bursaphelenchus xylophilus, the pinewood nematode responsible for pine wilt disease (Kikuchi et al., 2009). Since then, numerous LAMP assays have been reported for economically important species, including Globodera pallida, G. rostochiensis, Meloidogyne incognita, M. mali, M. hapla, M. chitwoodi, M. fallax, M. partityla, Radopholus similis, Tylenchulus semipenetrans, and Anguina wevelli (Ahuja et al., 2021; Camacho et al., 2024; Yang et al., 2024; Yu et al., 2018; Zhang and Gleason, 2019; Zhou et al., 2017). With the expanding use of LAMP in nematode diagnostics, developing and validating an assay targeting G. ellingtonae would be useful particularly in regions where G. ellingtonae co-occurs with the regulated Globodera species, G. pallida and G. rostochiensis, and where rapid, species-specific identification is essential to support quarantine decisions and management responses.
In this study, a species-specific LAMP assay was developed targeting the chorismate mutase (cm) gene to enable rapid and accurate identification of G. ellingtonae. The cm gene shows clear species-level sequence divergence and alternative splicing patterns among Globodera spp., making it an attractive diagnostic marker (Chronis et al., 2014; Yu et al., 2011). Exploiting these differences, the LAMP assay was designed and validated to reliably distinguish G. ellingtonae from the regulated PCN species G. pallida and G. rostochiensis, including in mixed-species samples where morphology-based identification is difficult. By providing a rapid, and sensitive method for species-level detection, this assay helps in diagnostics, and regulatory and management program aimed to safeguard potato production where Globodera spp. might co-exists.
A Globodera ellingtonae population used in this study originated from a potato field near Powell Butte, Oregon, USA, and was obtained from Dr. Inga Zasada (USDA-ARS). Nematodes were reared on the susceptible potato cultivars “Desiree” or “Russet Burbank” in a greenhouse under standard conditions (18℃ ± 2℃ daytime, 10℃ ± 2℃ night-time, 16:8 h light:dark photoperiod) for 12 weeks. Cysts were extracted from soil using an elutriator, dried, and stored at 4℃ until use (Dandurand and Knudsen, 2016).
For DNA extraction, single cysts were surface sterilized with 0.3% sodium hypochlorite for 5 min, followed by five rinses in sterile water (Nour et al., 2003). Cysts were hydrated in 0.5% gentamicin for 48 h and then crushed to release eggs. After adding 100 µL of potato root exudates, eggs were incubated at 18℃ ± 2℃ for 2 weeks to obtain pre-parasitic second-stage juveniles (J2s), which were collected from single cyst using a modified protocol based on (Subbotin et al., 2020). Genomic DNA (gDNA) was isolated from the collected J2s. J2s were lysed in 200 µL of nematode lysis buffer (250 mM NaCl, 200 mM Tris-HCl pH 8.5, 25 mM EDTA, 0.5% sodium dodecyl sulfate, 120 µg mL⁻¹ proteinase K) mixed with 75 µL of 3 M sodium acetate (pH 5.2) and 0.2 mL of 1 mm acid-washed zirconium beads (OPS Diagnostics). Homogenization was performed using a mini bead-beater (BioSpec 3110Bc) at 4,800 oscillations min⁻¹ for 20 s, and repeated three times. Samples were frozen at −20℃ for 20 min, then centrifuged at 13,000 × g for 10 min at 4℃. DNA was precipitated with isopropanol, centrifuged for 30 min, and pellets were washed sequentially with 70 and 100% ethanol. The dried DNA pellet was resuspended in 30–50 µL of TE buffer (10 mM Tris-HCl, 0.5 mM EDTA, pH 8.0). DNA purity and concentration were assessed using a NanoDrop ND-2000c spectrophotometer, and samples were stored at −20℃ until use (Solo et al., 2021).
LAMP primers specific to G. ellingtonae were designed based on the cm gene. A consensus alignment of cm sequences from G. ellingtonae and related cyst nematodes was generated in Bio Edit to identify regions unique to G. ellingtonae while avoiding conserved motifs that could cross-amplify non-target species. The alignment included sequences from G. ellingtonae (KF360245, KF360243), G. pallida (AJ487621, HM148925), G. rostochiensis (EF437152, EF437153), G. tabacum (HM148920, HM148922), Heterodera schachtii (DQ176601), and H. glycines (MG871363) (Fig. S1). Primer sets were designed using Primer Explorer V5 (https://primerexplorer.eiken.co.jp/lampv5e/index.html), with parameters adjusted to ensure species specificity and optimal thermodynamic properties.
For G. ellingtonae, five primers were designed: outer primers F3 and B3, inner primers FIP and BIP, and loop primers LF. FIP consisted of the F1c (3′ complementary) and F2 (5′) regions fused in tandem, whereas BIP combined the B1c (3′ complementary) and B2 (5′) regions. This configuration enables efficient strand displacement and exponential isothermal amplification. Primer sequences, Tm, length, and target region length are summarized in Table 1. Additionally, primer sets were designed for G. pallida and G. rostochiensis to evaluate assay specificity. Regions prone to nonspecific binding or secondary structure were avoided to minimize cross-amplification. All primers were synthesized by Sigma-Aldrich (USA).
Oligonucleotide sequences of LAMP primers targeting the cm gene for Globodera ellingtonae, Globodera rostochiensis, and Globodera pallida.
| Species | Primer | Type | Sequence (5′–3′) | Length (bp) | Tm | Target region length (bp) |
|---|---|---|---|---|---|---|
| Globodera ellingtonae | Ge-F3 | Forward outer | CCAGCAGATTCTTTTTTACATCA | 23 | 57.08 | ∼315 |
| Ge-B3 | Backward outer | AGTCGGCTTACAACACAT | 18 | 55.37 | ||
| Ge-FIP | Forward inner | ATTGCGCAAAATGATTTCCTCCGAAT ACTTCCCAAAAGATTACCTAAC | 48 | |||
| Ge-BIP | Backward inner | TGTGCCTTCATGAAGAACATTGATCCGTTGTCGATTTCGTT | 41 | |||
| Ge-LP | Loop | TCAAATTTGTCGTTGGGATGGAAGG | 25 | |||
| Globodera rostochiensis | Gr-F3 | Forward outer | TCTTCATTGTCGGCATGG | 18 | 56.54 | ∼297 |
| Gr-B3 | Backward outer | ACCTTTTTTACCTGAATGACTT | 22 | 55.59 | ||
| Gr-FIP | Forward inner | CAACCTTTTCCCGCTCGAAATCGTTG GCCAAAGATGTGGT | 40 | |||
| Gr-BIP | Backward inner | GCGAAGAGTGCCGGGATAAGAGCGTCCATTTGGTCTTG | 38 | |||
| Gr-LP | Loop | CAACTACGGGGAGCCGTTTT | 20 | |||
| Globodera pallida | Gp-F3 | Forward outer | TTTCCAAAAGATTGCCTAGC | 20 | 55.74 | ∼285 |
| Gp-B3 | Backward outer | CGTTTAGTGACCGTACCT | 18 | 55.06 | ||
| Gp-FIP | Forward inner | CAATGTTCTTCATGAAGGCACAGTCACAACAAATGCAAGTCATCG | 45 | |||
| Gp-BIP | Backward inner | CCAAACGGAAACGGAATCCCTGATTTGGCTTACAACACAT | 40 | |||
| Gp-LP | Loop | GGACTTGCGCAAAATGATTTCCTC | 24 |
LAMP reactions were performed with final volume of 25 µL, containing 1.5 µL of 10× isothermal amplification buffer, 3 µL of 10 mM dNTP mix, 1.5 µL each of FIP and BIP (30 pmol), 1 µL each of F3, B3, and loop primers (10 pmol), 1 µL of Bst DNA polymerase (1,200 U mL⁻¹; Invitrogen, Thermo Fisher Scientific), 2 µL of MgSO₄ (100 mM), 1 µL of template gDNA (1 ng), 4.8 µL of betaine (1.6 M), and 6.7 µL of nuclease-free water. To optimize amplification conditions, reactions were incubated at 56–66℃ (56, 58, 60, 62, 64, and 66℃) for 30, 45, 60, 75, or 90 min. Reactions were terminated by heating at 80℃ for 5 min.
For close-tube colorimetric detection, 2 µL of SYBR Green I nucleic acid stain (Invitrogen, Thermo Fisher Scientific) was pre-spotted onto the inner surface of each tube cap on a small piece of parafilm, as mentioned by Sukphattanaudomchoke et al. (2020). After amplification, tubes were briefly centrifuged to mix the stain with the amplified reaction, and products were evaluated by visual color change under ambient or blue light. LAMP products were further confirmed by electrophoresis on 1.5% agarose gels stained with SYBR Safe DNA gel stain and visualized using an Azure 200 system. Each reaction was performed in triplicate, and the full optimization experiment was repeated.
The cm gene of Globodera spp. were amplified using Bio-Rad C1000 thermal cycler with gDNA of G. ellingtonae, G. pallida, and G. rostochiensis. The PCR and qPCR were performed using the primer pair of F3/B3 designed in this study. PCR reactions were performed with final volume of 12.5 µL, containing 1 µL gDNA (1 ng), 1 µL each of forward and reverse primers (10 pmol; F3 and B3, Table 1), 1 µL of Taq DNA polymerase, 2 µL of reaction buffer, and 1 µL of 2.5 mM dNTP mix. PCR amplification was carried out by following one cycle of initial denaturation at 95℃ for 1 min; followed by 40 cycles amplification at 95℃ for 20 s, 62℃ for 15 s, and 72℃ for 30 s; and a final extension at 72℃ for 10 min. The PCR products were resolved on 1.5% agarose gels stained with SYBR Safe DNA gel stain (Invitrogen, Thermo Fisher Scientific) and visualized using an Azure 200 imaging system.
The real-time PCR amplification was performed as described by PCR amplification. The cm gene of G. ellingtonae was amplified using qTower iris (Analytical Jena). The standard curve for absolute quantification was generated using a tenfold serial dilutions series of G. ellingtonae gDNA, ranging from 1 ng to 10 fg (1 ng, 100 pg, 10 pg, 1 pg, 100 fg, and 10 fg). The real-time PCR profiled consisted of an initial denaturation at 95℃ for 1 min, followed with 40 cycles of amplification at 95℃ for 15 s, annealing at 62℃ for 30 s, and extension at 72℃ for 15 s. Data were analyzed using qPCRsoft (Analytik Jena) to determine cycle threshold (Cq) values; concentrations were calculated via linear regression of the standard curve. Melting curve analysis was performed by increasing the temperature of the sample reaction from 60℃ to 95℃ increments of 0.5℃ continuously at 0.05 s. Each reaction was performed in triplicates; assay was repeated thrice.
To determine the analytical sensitivity of the LAMP assay, a tenfold serial dilutions series of G. ellingtonae gDNA, with the initial concentration at 1 ng µL−1 (1 ng, 100 pg, 10 pg, 1 pg, 100 fg, and 10 fg) were prepared. All dilutions were evaluated in triplicate, and the assay was repeated to confirm reproducibility.
To confirm the specificity of the LAMP primer for G. ellingtonae and to ensure the absence of cross-amplification, reactions were performed using gDNA of G. pallida and G. rostochiensis as non-target species. gDNA of G. ellingtonae was used as the positive control, while nuclease-free water served as the non-template negative control. Genomic DNA from all the species were extracted using the lysis-buffer method as described above. Each 25 µL reaction contained 1.5 µL each of FIP and BIP (30 pmol), 1 µL each of F3, B3, and loop primers (10 pmol), 1 µL of template gDNA (1 ng), and the previously optimized reaction components. Reactions were incubated at the selected optimal temperature (e.g., 62℃) for 30 min in a water bath and terminated at 80℃ for 5 min. Real-time PCR specificity was evaluated using gDNA from G. ellingtonae, G. pallida, and G. rostochiensis with G. ellingtonae specific primers. All reactions were performed in triplicate, and the specificity assay was independently repeated to confirm reproducibility.
LAMP products were assessed by close-tube SYBR Green I stain and visual inspection of color and fluorescence, and by 1.5% agarose gel electrophoresis stained with SYBR Safe. Only G. ellingtonae DNA consistently produced the expected amplification signal, whereas no amplification was observed for non-target nematodes or negative controls in colorimetric observation as well in real-time PCR, confirming the specificity of the assay.
For G. ellingtonae, PCR amplification of the cm gene produced a single, clear amplicon of 310 bp, distinct from the products obtained from other Globodera species (Fig. 1a). Species-specific amplification obtained with the F3 and B3 primers, together with qPCR validation, confirmed the specificity of the cm gene region for distinguishing G. ellingtonae, from G. pallida and G. rostochiensis, supporting its suitability as a diagnostic molecular target for development of the LAMP assay (Fig. 1a and b).
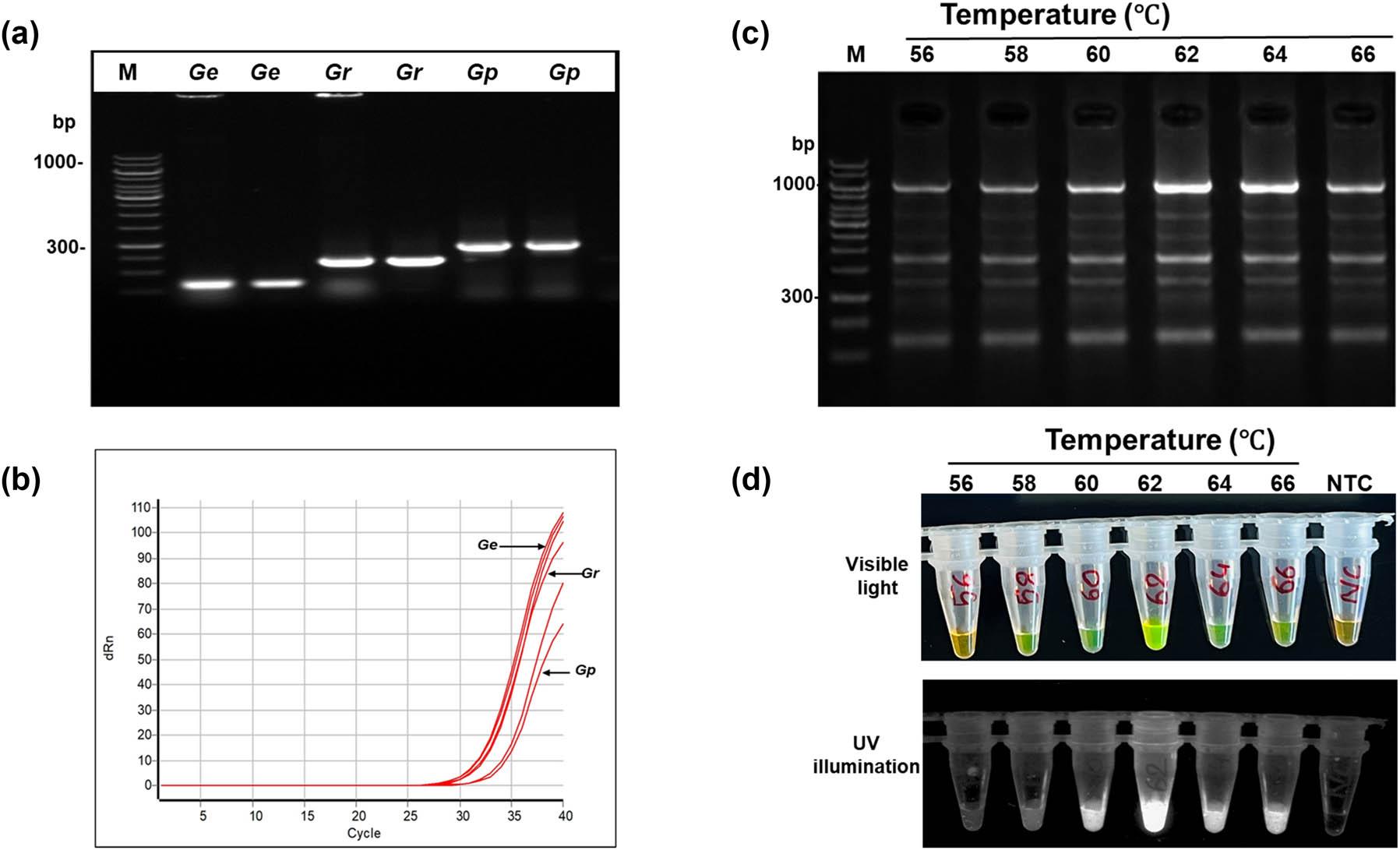
Optimization of LAMP assay for Globodera ellingtonae. (a) amplification of the cm gene using species-specific LAMP outer primers (F3 and B3), resolved on 1.5% agarose gel. Lane:100 bp DNA ladder; Lanes 2 and 3: Globodera ellingtonae (Ge); lanes 4 and 5: Globodera rostochiensis (Gr); and lanes 6 and 7: Globodera pallida (Gp). (b) qPCR amplification curves for the cm gene of Globodera ellingtonae (Ge); Globodera rostochiensis (Gr); and Globodera pallida (Gp). (c) Optimization of LAMP amplification temperature from 56 to 66℃, analyzed on 1.5% agarose gel. The LAMP assay conduced at 62℃ produced the brightest bend on the gel. (d) Colorimetric closed tube LAMP assay at the amplification temperature from 56 to 66℃ visualized under visible light (top) and UV illumination (bottom). Here, positive reactions appear green, whereas negative reactions and non-template control (NTC) remain orange in color due to staining with SYBR Green I dye.
LAMP amplification conditions were optimized using G. ellingtonae gDNA. The optimal amplification temperature and time were 60℃ for 30 min, followed by a termination at 80℃ for 5 min. Under these conditions, G. ellingtonae produced strong positive reactions, characterized by a visible color change from colorless to green fluorescence and the characteristic ladder‑like banding pattern on agarose gels (Figs 1c, d and 2a, b). Also, LAMP assays have been developed for all three Globodera species (Table 2). All subsequent LAMP experiments for G. ellingtonae detection were performed using these optimized conditions (Fig. 2c).
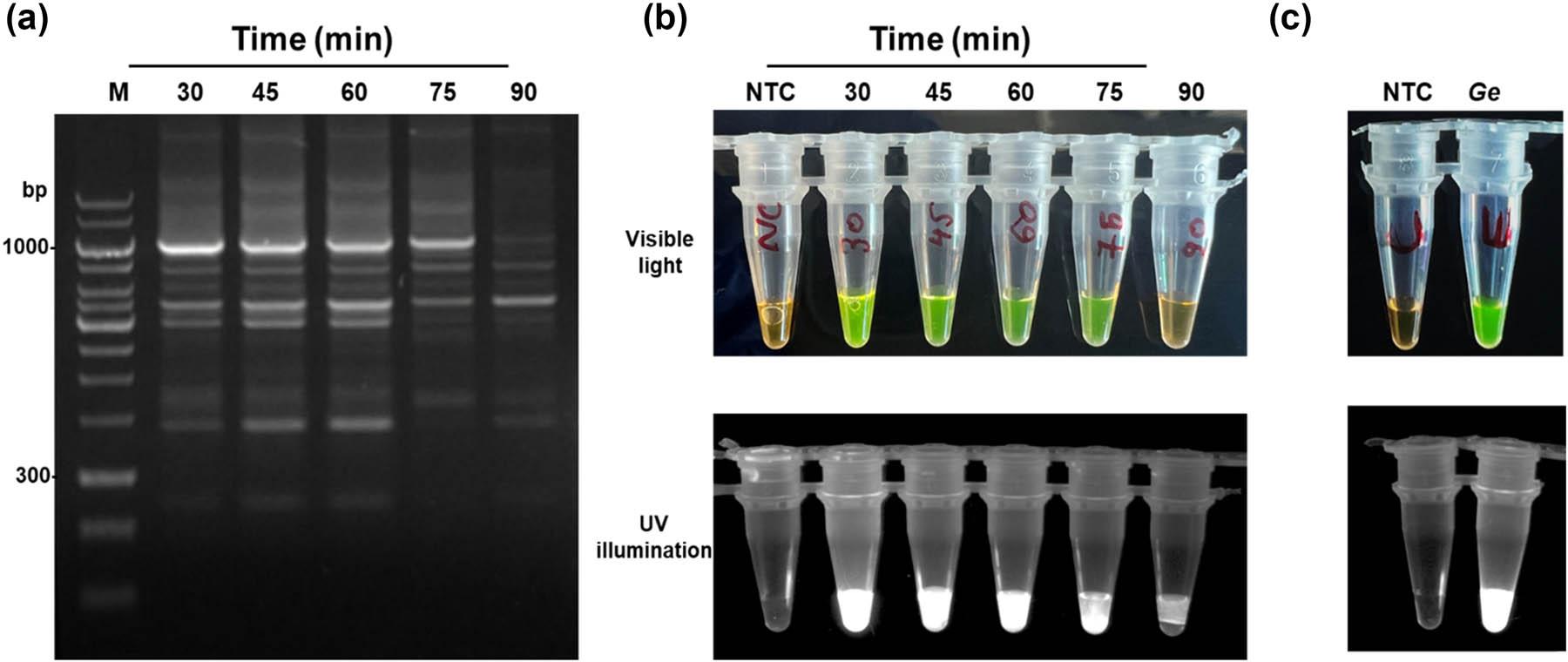
Optimization of LAMP assay for Globodera ellingtonae. (a) Optimization of LAMP amplification time from 30 to 90 min, analyzed on 1.5% agarose gel. The LAMP assay conducted at 30 min produced the brightest bend on the gel. (b) Colorimetric closed tube LAMP assay at the amplification temperature from 30 to 90 min visualized under visible light (top) and UV illumination (bottom). (c) Optimized LAMP reaction compared with NTC under visible light (top) and UV illumination (bottom). Here, positive reactions appear green, whereas negative reactions and NTC remain orange in color due to staining with SYBR Green I dye.
Optimized LAMP reaction parameters for Globodera pallida, Globodera rostochiensis, and Globodera ellingtonae
| Optimization | Optimized LAMP reaction for G. pallida | Optimized LAMP reaction for G. rostochiensis | Optimized LAMP reaction for G. ellingtonae | |
|---|---|---|---|---|
| dNTPs (mM L−1) | 0.2–1.8 | 2.1 | 2.1 | 2.1 |
| MgSO4 (mM L−1) | 2–10 | 1.2 | 1.4 | 1.4 |
| Bst DNA Polymerase (U µL−1) | 0.04–0.12 | 0.08 | 0.08 | 0.08 |
| Temperature (℃) | 54–66 | 60 | 62 | 62 |
The analytical sensitivity of the LAMP assay was determined using tenfold serial dilutions of G. ellingtonae gDNA, starting from 1 ng to 10 fg. Consistent amplification was observed down to 100 fg (Fig. 3a) in agarose gel and colorimetric and fluorescent dye indicator (Fig. 3b). While real-time PCR showed positive amplification to 1 pg (Fig. 3c) and conventional PCR showed lowest sensitivity with 10 pg. Compared to these standard methods, the developed LAMP assay showed a 100‑fold improvement in sensitivity over conventional PCR. The results showed that the assay is well suited for detecting low template quantities expected in regulatory and field samples.
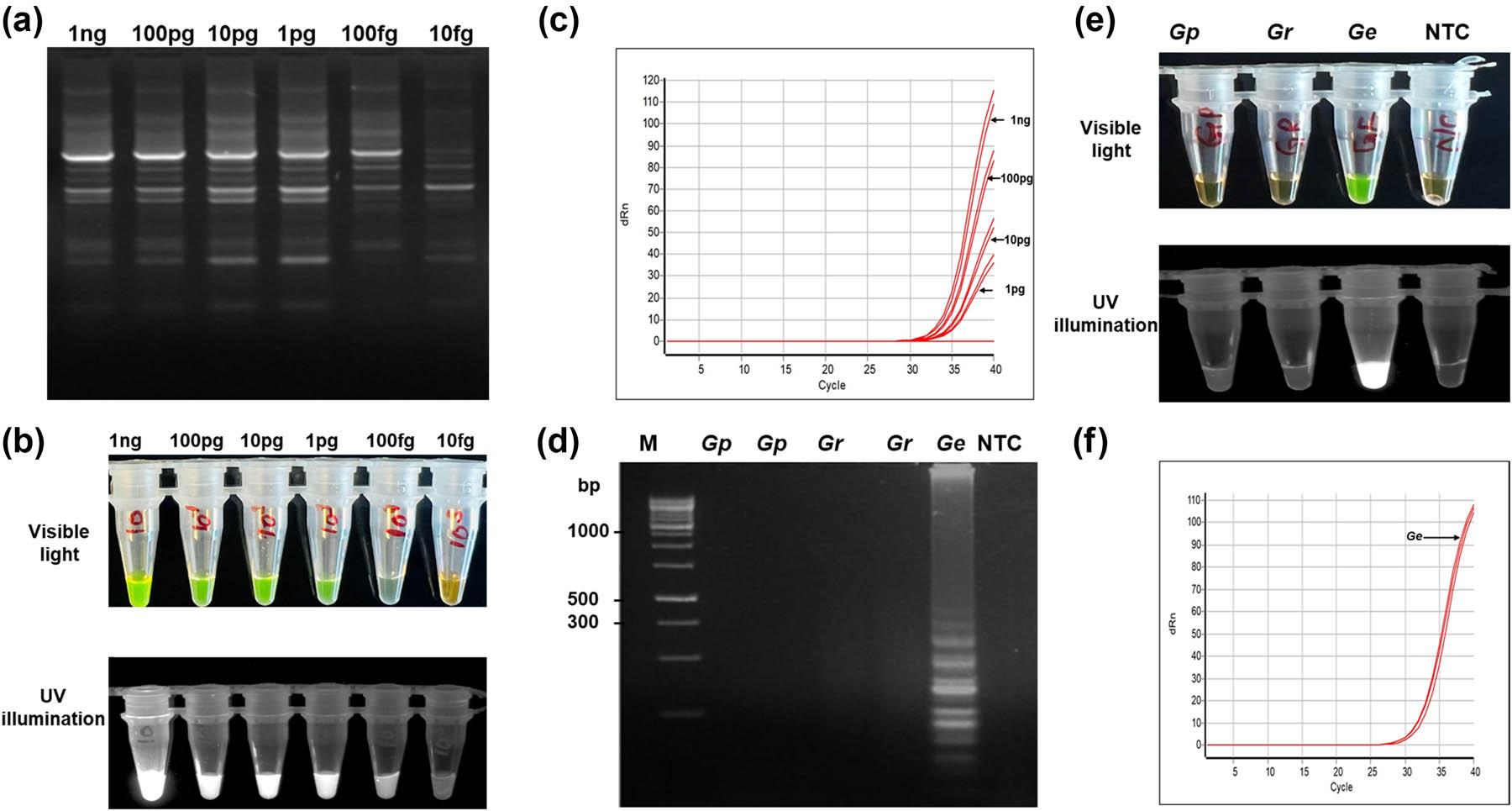
Specificity and sensitivity of the close-tube LAMP assay for G. ellingtonae. (a) Sensitivity of the LAMP assay using serially tenfold diluted gDNA of G. ellingtonae starting from 1 ng to 10 fg, visualized by 1.5% agarose gel electrophoresis. Each reaction contained 1 µL of template DNA per assay. The LAMP assay consistently detected till 100 fg. (b) Colorimetric closed tube LAMP assay visualized under visible light (top) and UV illumination (bottom). Here, positive reactions appear green, whereas negative reactions remain orange in color due to staining with SYBR Green I dye (c) qPCR amplification of serially diluted G. ellingtonae gDNA. (d) Specificity of the LAMP assay using G. ellingtonae specific primers evaluated against gDNA from G. pallida (Gp), G. rostochiensis (Gr), G. ellingtonae (Ge), and NTC. Amplification was resolved in 1.5% agarose gel. (e) Colorimetric closed tube LAMP assay visualized under visible light (top) and UV illumination (bottom). (f) qPCR specificity assay performed using G. ellingtonae specific primers with gDNA from G. ellingtonae, G. pallida, and G. rostochiensis.
Species specificity was evaluated using G. ellingtonae DNA with other Globodera species. Only reactions containing G. ellingtonae DNA produced a positive signal, as evidenced by green fluorescence and the ladder‑like banding pattern on agarose gels (Fig. 3d), and colorimetric in closed tube (Fig. 3e) and in real-time PCR (Fig. 3f). No amplification, including late or weak signals, was observed for non‑target DNA or no‑template controls (Fig. 3d and e). These results demonstrate that the cm‑based LAMP assay is highly specific for G. ellingtonae and suitable for differential diagnosis in samples where closely related Globodera species may co‑occur.
The PCNs G. pallida and G. rostochiensis are economically important nematode pests of potato due to their ability to cause severe yield losses. In regions such as Idaho (USA), Chile, and Argentina, the presence of G. pallida alongside the non-regulated species G. ellingtonae further complicates detection and species-level diagnosis (Hesse et al., 2021; Lax et al., 2014; Phillips et al., 2017). Mixed populations of Globodera species with G. ellingtonae further increase the diagnostic challenges. Accurate and timed identification is essential for effective pest management and phytosanitary decision-making. Traditional identification of G. pallida and G. rostochiensis has relied on morphological examination of cysts and second-stage juveniles (J2s), focusing on physical traits like cyst shape, size, and color; J2s body and tail morphology; stylet length and structure; and vulval basin characteristics (Nisa et al., 2022; Skantar et al., 2011; Wainer and Dinh, 2021; Xu et al., 2023). Although these methods are foundational in nematology, taxonomical identification demands specialized in-depth taxonomic expertise which is time-consuming. To overcome these limitations, molecular diagnostic tools such as isoenzyme electrophoresis, PCR, multiplex PCR, RFLP, and qPCR have been developed (Nisa et al., 2022; Subbotin and Franco, 2022; Vallejo et al., 2021). Although these methods are practical and accurate, they depend on costly reagents, sophisticated laboratory infrastructure, and trained personnel, which may restrict their routine use.
In this study, a species-specific, fluorescent, and colorimetric closed-tube LAMP assay was developed to distinguish G. ellingtonae from G. pallida and G. rostochiensis. The assay targets the (cm) gene, a well-characterized effector gene involved in host–pathogen interactions (Chronis et al., 2014; Yu et al., 2011). A semi-layer of parafilm was used in a closed-tube format to separate the SYBR Green I dye from the reaction mixture during amplification and allow post-amplification staining without opening the tube (Sukphattanaudomchoke et al., 2020). The presence of ladder-like bands on gel electrophoresis (Figs 1 and 2) and amplification in real-time PCR confirmed the successful amplification of the LAMP assay. The colorimetric and fluorescent dye indicators were used to interpret the result without the post-amplification preparation and minimize the risk of cross-contamination during post-amplification handling (Figs 1d, 2b, and 2c).
The cm gene is broadly conserved across plant-parasitic nematodes but exhibits species-specific sequence variation and splicing patterns, making it a suitable marker for molecular diagnostics (Bekal et al., 2003; Yu et al., 2011) (Fig. S1). Previous studies have used cm-based assays, including TaqMan qPCR and multiplex PCR, for G. pallida identification; however, the reports focused on G. pallida, with limited evaluation of specificity across closely related species (Bekal et al., 2003; Chronis et al., 2014). The LAMP assay described here is, to our knowledge, the first to incorporate all three Globodera species in a comparative specificity framework, explicitly including G. ellingtonae, which has not been targeted previously by LAMP-based diagnostics (Fig. 3d).
The developed LAMP assay showed high sensitivity, reliably detecting G. ellingtonae gDNA at concentrations as low as 100 fg (Fig. 3a), whereas conventional cm-based PCR was limited to 10 pg (Fig. 3c). This 100-fold improvement in sensitivity is consistent with previous LAMP studies for other plant-parasitic nematodes, including Meloidogyne chitwoodi, M. fallax, M. incognita, and Radopholus similis, which reported detection limits around 100 fg µL⁻¹ (Camacho et al., 2024; Niu et al., 2011; Suwanngam et al., 2025; Zhang and Gleason, 2019; Zhou et al., 2017). Importantly, the present assay achieves comparable or better sensitivity while being validated against closely related Globodera species, demonstrating both diagnostic utility and robustness.
In terms of specificity, the assay showed no cross-reactivity with G. rostochiensis or G. pallida when evaluated using G. ellingtonae-specific primers (Fig. 3d). This high specificity is critical for molecular diagnostics, as non-specific amplification can generate false positives and undermine confidence in assay results. The lack of non-target amplification suggests that careful primer design successfully mitigated common issues such as primer–dimer formation and off-target binding, which frequently complicate LAMP assay development (Ding et al., 2019; Zhang and Gleason, 2019; Zhou et al., 2017). The ability to detect DNA from a single cyst also increases the assay’s utility for early detection and use in mixed populations, where accurate species identification underpins timely regulatory action. The overall performance aligns well with other LAMP-based diagnostics for plant-parasitic nematodes and reinforces its robustness for practical application (Bairwa et al., 2023; Camacho et al., 2024).
A major advantage of the developed assay is its compatibility with low-cost equipment, such as a simple heating block or water bath, eliminating the need for a thermocycler or real-time PCR. This feature is especially valuable for survey programs, and regional laboratories where rapid and sensitive diagnostics are essential, but resources are limited. Previous work has emphasized the importance of cost-effective molecular tools for plant biosecurity and pathogen surveillance, and this assay directly addresses those needs (Ahuja and Somvanshi, 2021; Notomi et al., 2015; Zhang and Gleason, 2019).
Given its short amplification time (<30 min), high sensitivity, and strong species specificity, the LAMP assay has clear potential for integration into mixed-population testing, particularly in regions where G. ellingtonae coexists with G. pallida and G. rostochiensis. In such contexts, traditional morphology-based approaches are challenging and slow, whereas a species-specific molecular assay enables rapid and precise discrimination. Future research should extend validation to additional G. ellingtonae populations and closely related Globodera species from diverse geographic regions (for example, South America, Europe, and other major potato-producing areas) and evaluate performance directly on field-derived samples and composite soil extractions.
In conclusion, this study presents the LAMP assay for G. ellingtonae and the comparative validation across the three major Globodera species affecting potato. The assay provides rapid, sensitive, and specific diagnostics method, which is well suited to implement in regulatory sites and in management programs. Developed LAMP assay showed a significant advancement in molecular assays for PCN management and phytosanitary regulation.
This work was supported by the USDA-SCRI award number “NIFA-SCRI 2022-51181-38450.”
The authors express their gratitude to Dr. Inga Zasada for providing Globodera ellingtonae and Dr. Xiaohong Wang for supplying Globodera rostochiensis. Additionally, the authors extend their appreciation to the PCN team at the University of Idaho for their valuable contributions to the project.
This work was supported by the USDA-SCRI Award number NIFA-SCRI 2022-51181-38450.
CS: Conceptualization, methodology, investigation, formal analysis, validation, visualization, writing original draft preparation, review and editing; LMD: Conceptualization, methodology, supervision, project administration, resources, funding acquisition, review and editing.
The authors declare that they have no conflict of interest.
The data supporting the findings of this study are available from the corresponding author upon reasonable request.