
The Qinghai-Tibet Plateau (QTP), recognized as the largest and highest plateau globally, is frequently termed the “Roof of the World” and the “Third Pole” (Deng et al. 2011). Its extreme environmental conditions have contributed to its remarkable species diversity (Wu et al. 2022). The primary habitat of bharal in China is predominantly located within the Qinghai-Tibet Plateau and its adjacent regions. The bharal, a medium-sized herbivore, primarily inhabits alpine meadows and high-elevation mountains on the Tibetan Plateau (above 4,000 m), with forest grasslands or bare rocks as their primary habitat types, which are far from water resources (Liu et al. 2018). During the 20th century, anthropogenic pressures such as overhunting and habitat degradation precipitated a severe population decline, leading to its classification as a second-class protected species under China’s wildlife conservation framework and an endangered status. Current IUCN Red List assessments (Harris 2014) estimate the wild adult population to range between 4,700 and 8,700 individuals (Jiang and Migmar 2020).
The abundance and diversity of viruses are remarkably high. Over recent decades, advancements in viral metagenomics have significantly enhanced our ability to identify viruses on various environmental surfaces. There is currently limited knowledge regarding the viromes in the feces of common mammals inhabiting the Qinghai-Tibet Plateau, although the fecal microbial community composition, diversity, as well as potential functions have been elucidated by using utilized 16S rRNA sequencing technology (Wang et al. 2022).
Astrovirus, a single-stranded positive-sense RNA virus, is recognized as a significant zoonotic pathogen capable of inducing diarrhea in humans and various other mammals (Rivera et al. 2010; Sajewicz-Krukowska and Domanska-Blicharz 2016; Vu et al. 2017). Recent investigations have established an association between astrovirus and fatal diarrhea in children under 5 years of age (Black et al. 2024). This virus exhibits substantial genetic variability and the capacity for recombination, enabling its continuous adaptation to new hosts and facilitating interspecies transmission (Wohlgemuth et al. 2019). However, the precise correlation between Mamastrovirus and specific diseases remains uncertain.
In this study, we collected feces samples from 10 wild bharal residing in the Chang Tang Nature Reserve, which is located in the heart of the Qinghai-Tibet Plateau. These samples were then subjected to virome analysis. A novel astrovirus was identified in the fecal samples, which is referred to as “QTPAstv” in the following text, and subsequent genetic evolution and recombination analyses were fully characterized. Additionally, we also acquired a novel virus genome showing a relationship to circoviruses and performed a phylogenetic analysis. In summary, our findings suggest the potential existence of novel viruses in wild Tibetan bharal inhabiting the Qinghai-Tibet Plateau region, thereby contributing to advancements in research on high-altitude wildlife mammals.
The present study undertook a viral metagenomic analysis by collecting and examining fecal samples from ten wild bharal in the Chang Tang Nature Reserve, which is located in the Qinghai-Tibet Plateau (QTP). All fecal samples were collected in sterile containers and stored on dry ice. Prior to constructing the viral metagenomic libraries, 0.5 ml of Dulbecco’s phosphate-buffered saline (DPBS) was prepared, and 10 g of each sample was added. The mixture was vortexed for 5 minutes and then incubated at 4°C for 30 minutes. Following this, samples were centrifuged at 15,000 × g for 10 minutes, and the supernatants were transferred to pre-labeled sterile 1.5 ml centrifuge tubes. Finally, the collected supernatants were stored at –80°C for future analysis (Lu et al. 2024). The sample collection process adhered strictly to the Wildlife Protection Law of the People’s Republic of China. Additionally, all experiments were performed in strict compliance with the guidelines outlined for a Biosafety Level 2 laboratory.
Individual fecal specimens (n=10) underwent standardized processing prior to NovaSeq sequencing and then mixed into a mixture and sequenced. Initially, 100 μl of supernatant was aliquoted from a previously prepared library sample and transferred into a sterile 1.5 ml centrifuge tube. Centrifugation was then performed at 12,000 × g for 5 minutes at 4°C to separate particles, followed by filtration through a 0.45 μm syringe filter to concentrate viral particles (Hoyles et al. 2014; Conceição-Neto et al. 2015). The resultant filtrates were subjected to enzymatic treatment with RNase and DNase to degrade unprotected nucleic acids, with digestion occurring at 37°C for 60 minutes (Zhang et al. 2017). Subsequently, the experimental procedure extracted total nucleic acids using the QiAamp viral RNA minikit. The viral nucleic acid samples were then reverse transcribed using the following enzymes: Super-Script™ IV Reverse Transcriptase (Invitrogen™, Thermo Fisher Scientific Inc., USA) and random hexamer primers. Following reverse transcription, a single cycle of DNA synthesis was conducted using Klenow fragment polymerase (New England BioLabs, USA). Then, libraries were prepared with the Nextera® XT DNA Library Preparation Kit (Illumina, Inc., USA), followed by sequencing on the Illumina®NovaSeq™ 6000 platform with 250 bp paired ends (Liu et al. 2016).
The 250-bp paired-end reads generated from sequencing were indexed using the software provided by Illumina. The data were subsequently processed through an in-house analysis pipeline running on a 32-node Linux cluster. Low-quality sequences and primer sequences at both ends were trimmed using Trim Galore v0.6.5. Contaminating sequences were further screened using the VecScreen tool (https://www.ncbi.nlm.nih.gov/tools/vecscreen). The resulting files underwent additional quality control using Trim Galore v0.6.5. For assembly and contig generation, the data were processed with the default parameters of MEGAHIT v1.2.978. To annotate the assembled sequences, the DIAMOND software blasts algorithm was used to compare each contig, and singlet reads against NCBI’s viral proteome database, which was downloaded in May 2023. A significance threshold of E-value less than 10−5 was applied for sequence selection. The filtered viral sequences were reanalyzed against an in-house non-viral, non-redundant (NVNR) protein database to further eliminate potential false-positive viral sequences. All steps were performed using the software’s default settings.
Using Megan v6.21.16 (Huson et al. 2016), the previously obtained reads were taxonomically classified into relevant categories. De novo assembly and reference-based mapping were then performed using Geneious Prime software version 2024.0 (Kearse et al. 2012) to assemble the reads and obtain a nearly complete virus genome. Nested PCR was employed for regions with gaps between contigs. The PCR reactions were prepared according to established protocols, and the pre-mixed Taq enzyme from Takara Biomedical Technology (China) will be added to the reaction system. The PCR cycling conditions were as follows: initial denaturation at 95°C for 5 minutes; 35 cycles of 95°C for 30 seconds, 50°C for 30 seconds, and 72°C for 45 seconds; followed by a final extension at 72°C for 10 minutes. All PCR reactions included negative controls, and amplified products were sequenced using the Sanger method. Additionally, Geneious prime v2019.0 (https://www.geneious.com) was utilized to predict and annotate open reading frames (ORFs) and design primers. Predicted ORFs were further analyzed using BLASTx for functional annotation. To address gaps in the 3’ terminal region, an expression vector was constructed and transfected into DH5α competent cells. Positive clones were identified using bacterial colony PCR and sequencing methods.
Phylogenetic analysis was performed using nucleotide or protein sequences of viruses identified in this study and related reference strains obtained from GenBank. Sequence alignment was conducted with the ClustalW algorithm in MEGA 10.1.8 (Kumar et al. 2018) under default parameters. The aligned sequences were then used to construct maximum likelihood trees through IQ-TREE (Minh et al. 2020). For Astroviridae, the phylogenetic tree was generated based on ORF1a (2,487 bp) and ORF2 (1,710 bp) nucleotide sequences. Circoviridae analysis utilized replication-associated protein (Rep) sequences. All trees were constructed using IQ-TREE with 1,000 ultrafast bootstrap replicates (-bb 1000) and the ModelFinder algorithm (-m MFP) to determine the optimal substitution model, which was identified as TIM2e+I+R4 through the software’s integrated ModelFinder program.
We utilized the Recombination Detection Program 4.39 (RDP4) (Martin et al. 2015) to verify the presence of recombination events within the viruses identified in our study. Initially, potential recombination events were identified by observing distinct clustering patterns formed by various regions of the viral genomes on phylogenetic trees. Based on these observations, viruses exhibiting potential recombination were selected for further investigation. The complete genome sequences of viruses suspected to undergo recombination were analyzed independently using RDP4 software to identify potential recombination events. Seven distinct algorithms within RDP4 were utilized for this analysis, including RDP, GENECONV, 3Seq, Chimaera, SiScan, MaxChi, and LARD. Recombination was considered to be true if detected by at least three of these methods, with a p-value cutoff of 0.05.
Throughout the entire experimental process, standard laboratory precautions were rigorously adhered to in order to minimize the risks of cross-contamination among samples and nucleic acid degradation. Specifically, aerosol filter tips were utilized to reduce the potential for sample cross-contamination. All materials that come into direct contact with nucleic acids were verified to be free of DNase and RNase activity. Samples were dissolved in diethylpyrocarbonate (DEPC)-treated water supplemented with RNase inhibitors. In this experiment, deionized water was utilized as a negative control sample. The negative control and experimental groups were subjected to further processing under identical experimental conditions. Analysis of the sequencing results revealed that the number of sequences generated by the negative control group was significantly lower, with no indication of viral contamination. These findings suggest that the methodology employed in this study effectively prevents potential viral contamination. Quality control measures were implemented throughout all aforementioned experimental procedures.
The library of bharal fecal samples sequenced on the NovaSeq platform generated 27,912,988 unique sequence reads. Through analysis and identification, 372,598 sequence reads were assigned to viruses. After de novo assembly, a total of 185,352 contigs were generated, among which 40 virus families were included. (Fig. 1) The following eight families exhibited a notably high number of sequences with potential to infect eukaryotic hosts: Circoviridae (n = 33), Herpesviridae (n = 20), Smacoviridae (n = 18), Baculoviridae (n = 18), Astroviridae (n = 13), Picornaviridae (n = 7), Retroviridae (n = 7), Alloherpesviridae (n = 4). This study primarily focused on the Astroviridae and Circoviridae families. Both viral families pose potential pathogenic risks to the bharal, and novel sequences were identified within each family. Notably, a recombination event was detected in the astrovirus, and the complete genome of the circovirus has been successfully obtained.
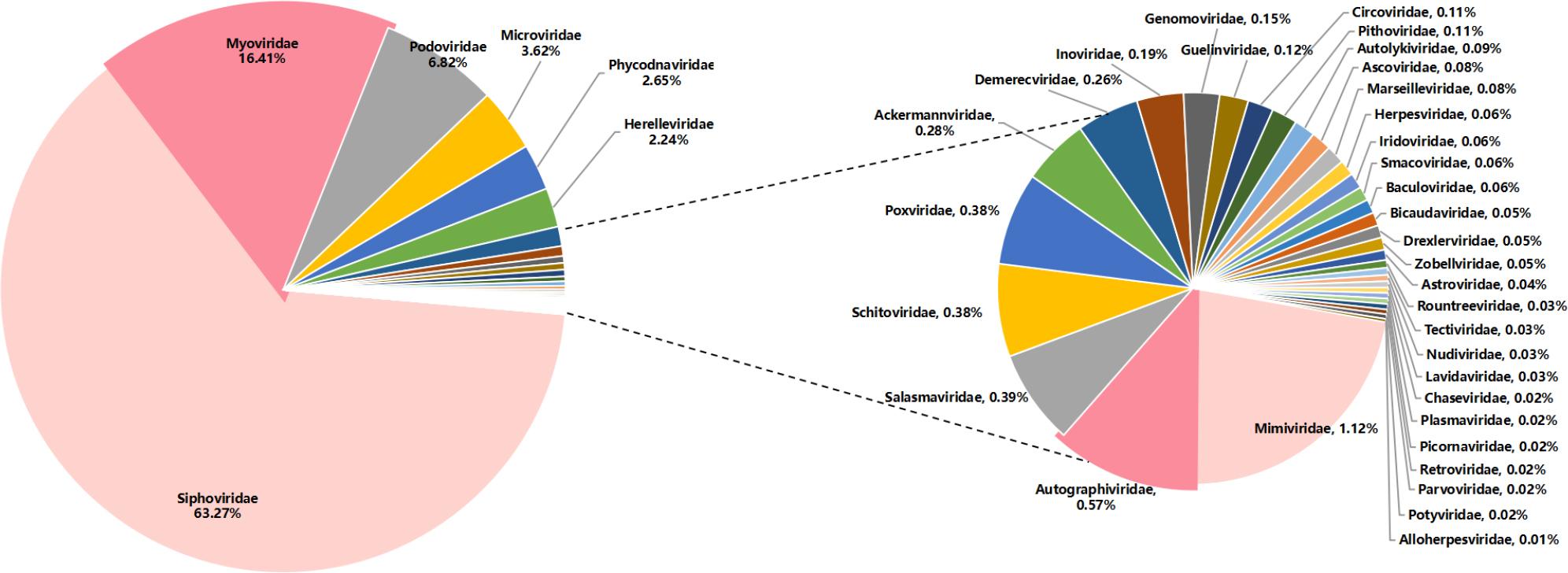
Viral composition of all libraries at the family level.
We successfully assembled the astrovirus, QTPAstv, resulting in a nearly complete genome sequence of the Astv. Additionally, we developed two primer sets specifically to amplify the missing 5’ and 3’ terminal sequences of the genome. The length of the QTPAstv genome is 6,032 base pairs, with the untranslated regions (UTRs) located at positions 1–346 and 5,979–6,032.
We identified three open reading frames (ORFs) within the nearly complete QTPAstv viral genome, which we designated ORF1a, ORF1b, and ORF2. There is a 7-nucleotide overlap between ORF1b and ORF2 (Fig. 2). Conserved domain analysis, conducted using the NCBI Conserved Domain Search tool, revealed that the conserved RdRp amino acid motif of QTPAstv is situated between positions 3,276 and 3,992 within the genome.
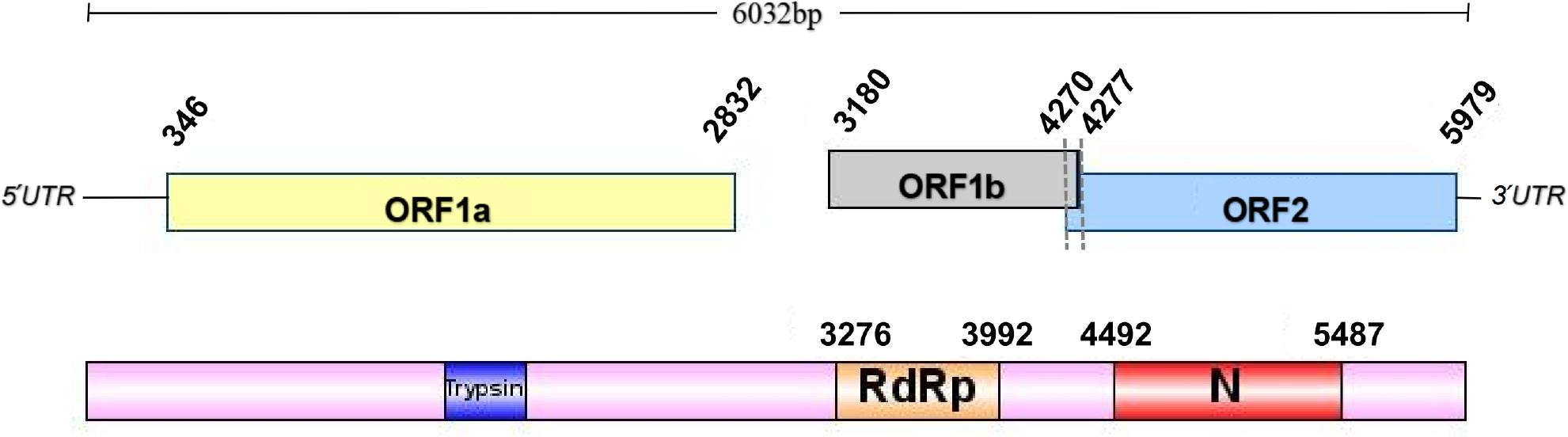
Genomic structure of QTPAstv. The conserved ribosomal frameshift site is marked.
In order to investigate the genetic relationship of QTPAstv, we analyzed the nucleotide sequences of ORF1a and ORF2 obtained from the dataset of this study. These sequences were then compared with reference sequences of mammalian astrovirus genotypes available in the GenBank database. As a result, two phylogenetic trees were constructed. (Fig. 3a and 3b) Sequence analysis using Blastp in NCBI indicated that the ORF1a of QTPAstv shares the highest amino acid sequence identity (93.48%) with the strain ORF1a from Henan mamastrovirus 4 (UMO75810), which was derived from the gut metagenome. In contrast, the ORF2 of QTPAstv shares the highest amino acid sequence identity (84.06%) with the capsid protein strain from Mamastrovirus 3 (XCY59703.1). Different phylogenetic trees were generated for various regions of the same virus, suggesting that QTPAstv may have undergone recombination.
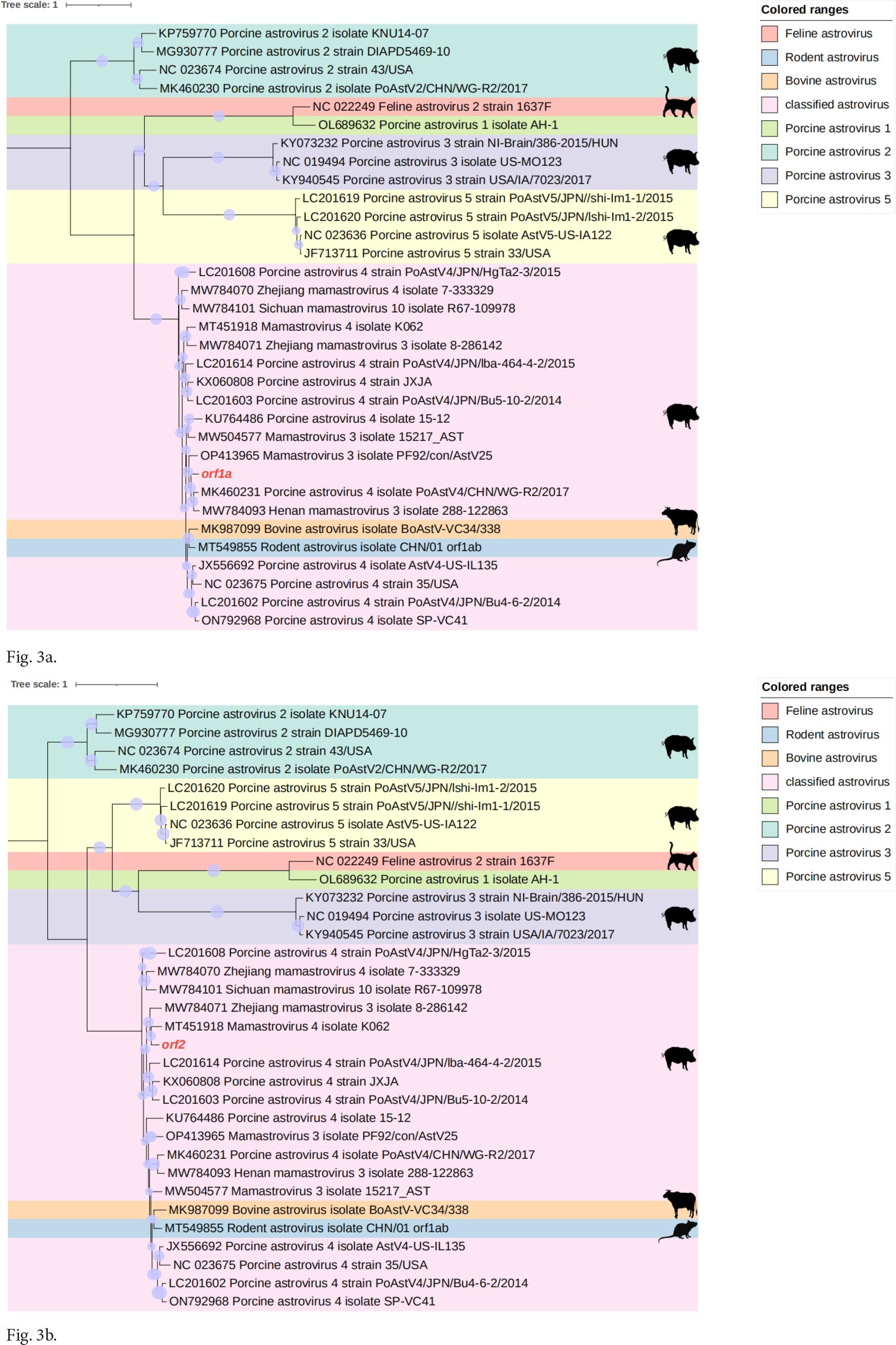
Phylogenetic analysis of QTPAstv. The maximum likelihood tree was constructed based on the nucleotide sequences of ORF1a and ORF2. Red branches represent the newly discovered viruses in this study.
Analysis using the seven recombination detection tools available in RDP 4.39 indicates that QTPAstv may be a recombinant virus, with OP413965 identified as its major parent and MW784071 as its minor parent (Fig. 4a). This result was supported by five algorithms (RDP, GENECONV, BootScan, MaxChi, Chimaera, 3Seq; Fig. 4b). We utilized four sequences to further extended: QTPAstv, the major parent and minor parent sequences derived from RDP analysis, and an outgroup sequence, which revealing significant sequence similarity between QT-PAstv and OP413965 in the 500–1510 nt region, as well as with MW784071 in the 4200–5900 nt region.
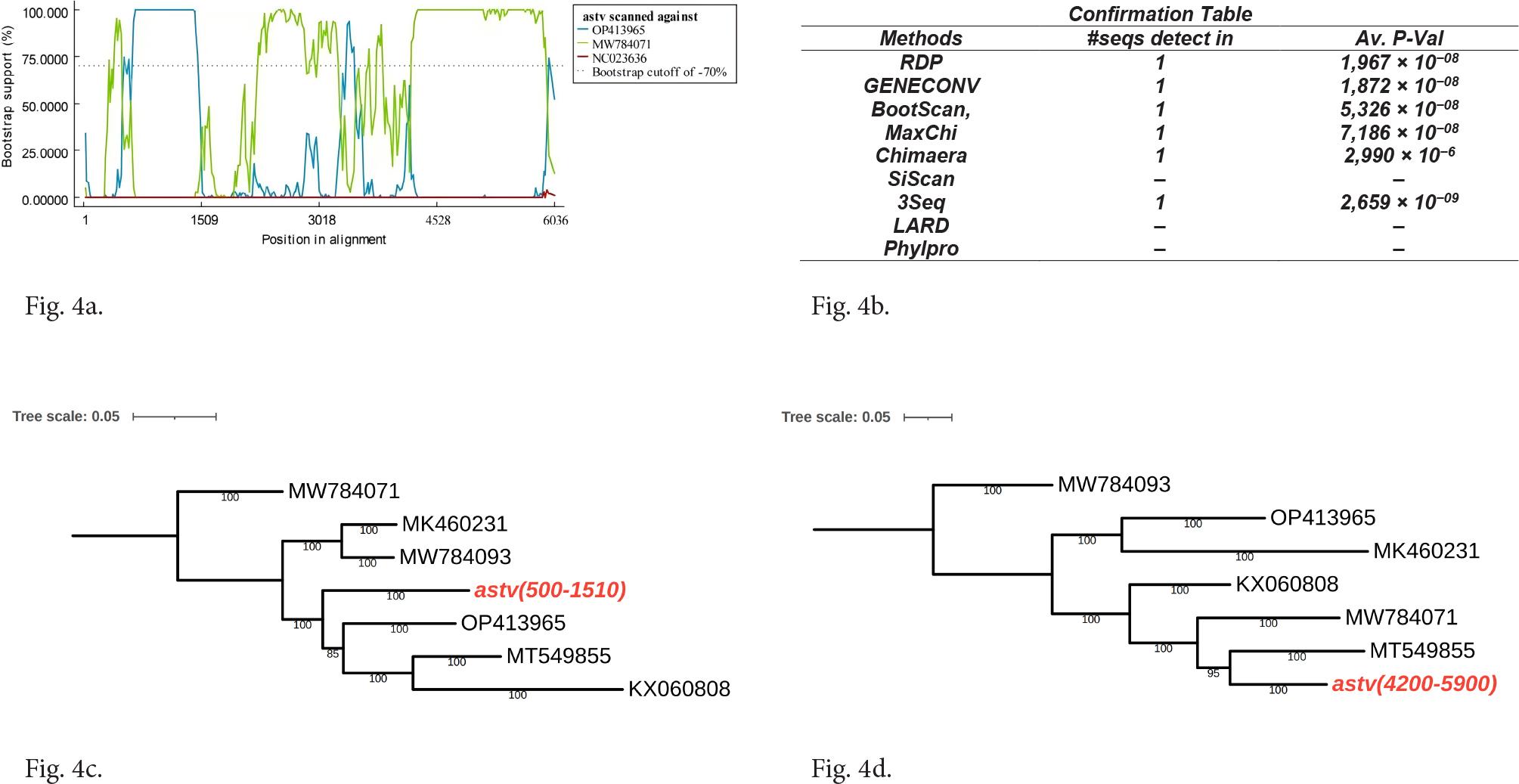
Recombination analysis of QTPAstv.
a) Manual BootScan methods on evidence of recombination events occurring with a window size of 200, a step size of 20, and a model of 100 Bootstrap replications. Manual BootScan methods selected QTPAstv as the query, NC023636 as the distantly related sequence, and OP413965 and MW784071 as the parents. b) The confirmation table shows the support obtained by the algorithms for the identified recombination events. c), d) Phylogenetic trees were constructed in the 500–1500 nt and 4200–5900 nt regions respectively.
To confirm the occurrence of the recombination event, a phylogenetic tree was constructed based on the regions adjacent to the recombination site. The tree constructed from sequence segments ranging from 500 to 1510 nt reveals that QTPAstv clusters with OP413965. In contrast, when sequences were divided into segments between 4200 and 5900 nt, QTPAstv was grouped with a clade containing MT549855 and MW784071 (Fig. 4c and 4d). These findings support the hypothesis that QTPAstv is a recombinant virus; however, the precise parental origin of the region between 1510 and 4200 nt remains unidentified.
Recent advancements in viral metagenomics have identified an increasing number of previously unknown viruses, particularly a significant abundance of uncharacterized Circoviridae viruses within animal gastrointestinal tracts. Additionally, single-stranded Circoviridae viruses, which encode circular replication (Rep) proteins, exhibit a global distribution and infect a diverse range of eukaryotic organisms.
We successfully assembled circovirus and obtained an almost complete genome sequence (Fig. 5a). Phylogenetic analysis based on rep protein construction showed that Circoviridae viruses demonstrate a broad host diversity, including arthropods, reptiles, birds, vertebrates, and unidentified hosts (Fig. 5b). Among them, in addition to environmental samples, the rep protein identified in this study showed the highest similarity to the AIY31238 protein sequences. The host of AIY31238 is the dromedary camel. These findings suggest that these viruses exhibit varying host preferences, indicating that the gut of the bharal in the QTP has unique ecological characteristics.
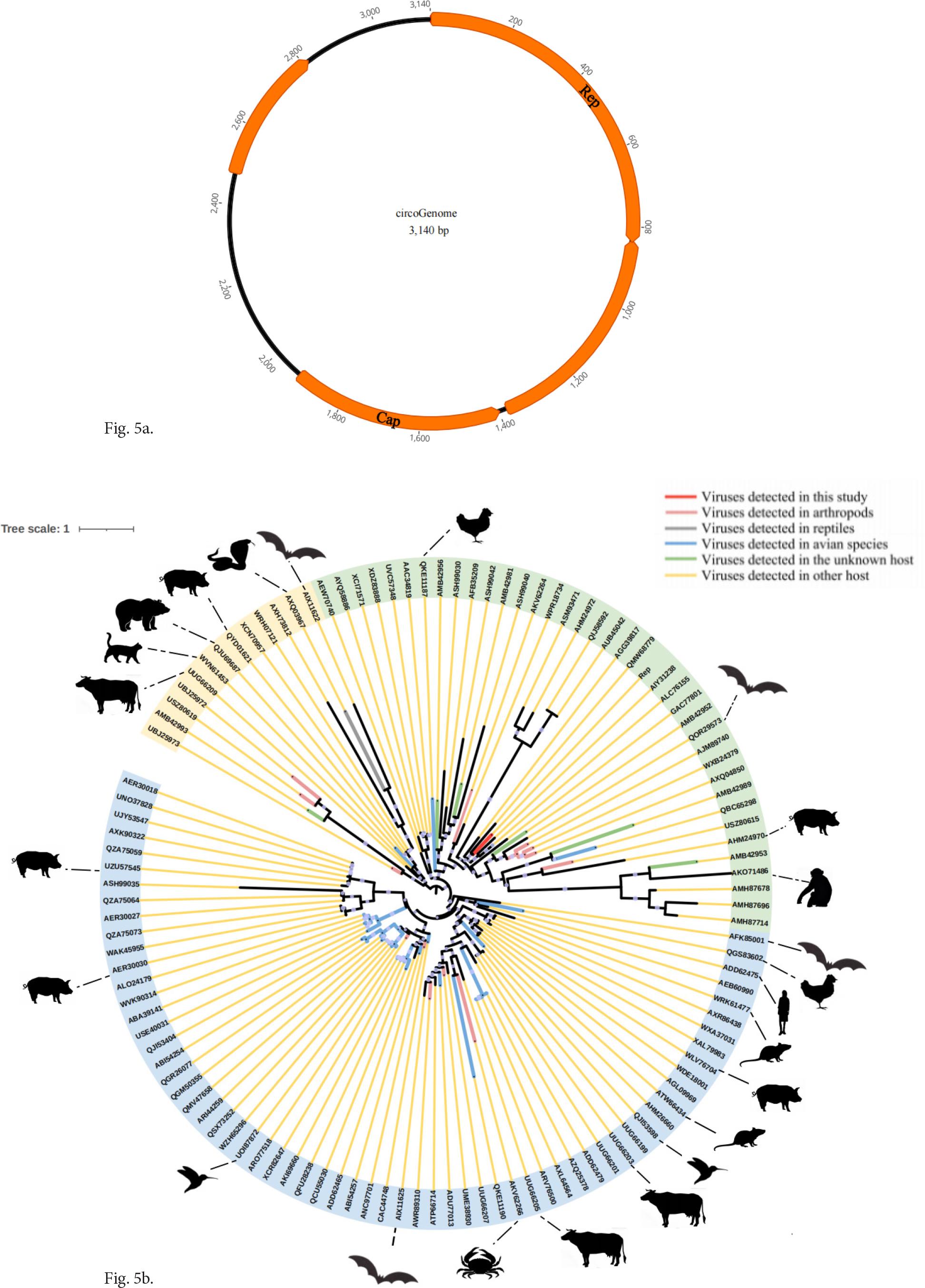
Novel circovirus identified in the feces of wild bharals.
a) Genome organization of circovirus. b) Identification and phylogenetic analysis of the Circoviridae family in the gut of the bharal. The Rep protein sequence of the Circoviridae family should be used to construct a maximum likelihood tree.
The comprehensive understanding of viral diversity in plateau mammals is crucial for studying virus evolution and expanding the repository of mammalian viruses. However, the viral diversity in apparently healthy wild mammals inhabiting the Chinese plateau remains largely uncharted (Yang et al. 2015). Recent metagenomic surveys have unveiled unprecedented viral diversity in wild bharal inhabiting highland regions (Jian et al. 2023; Liu et al. 2024). In order to gain a more comprehensive understanding of the presence of viruses in wild Bharal on the Qinghai-Tibet Plateau in China, we conducted a metagenomic analysis of fecal samples from the Chang Tang Nature Reserve and discovered a novel circovirus genome. Additionally, we investigated the diversity of this virus within the Circoviridae family.
Despite its widespread distribution, the astrovirus remains poorly understood in terms of diversity. Previous studies consistently reported ongoing recombination events of Mamastrovirus globally (Tse et al. 2011; Wohlgemuth et al. 2019; Hu et al. 2023). Identifying the novel viral genome QTPAstv in this study suggests that recombination events among astrovirus genomes may also be prevalent in wild mammals inhabiting the QTP. Given their potential to cause zoonotic diseases, further investigation should be conducted on their proteins’ higher-order structure and functionality to assess their risk for human infection. However, uncertainty remains regarding the susceptibility of wild mammals on the plateau to the disease and whether the collected fecal samples originated from healthy bharal.
The family Circoviridae currently includes 149 recognized species. Despite the lack of definitive evidence linking circovirus infection to specific diseases, certain species have been detected in humans with conditions such as paralysis (Smits et al. 2013), pneumonia (Prades et al. 2021), encephalitis, and diarrhea (Phan et al. 2015). Our study demonstrates that circovirus within the study cluster with AIY31238 has a camel as its host in addition to environmental samples of unknown origin. Furthermore, circoviruses associated with human diseases belong to distinct phylogenetic clades. This result suggests a specific correlation between the virus sequences of herbivores while highlighting key areas for future research to investigate circovirus-disease associations.
In conclusion, we have identified a novel astrovirus in wild bharal at the QTP and characterized its nearly complete genome. Recombinant analysis indicated that it was a recombinant with multiple recombinant events. Our research emphasizes the significant diversity of bharal viral communities in high-altitude regions, enriching these areas’ existing mammalian virus database. However, further studies are needed to uncover the role of bharal viruses in potential pathogenic mechanisms for both animals and humans.