
Myocardial infarction (MI) is a leading cause of morbidity and mortality worldwide, traditionally associated with obstructive coronary artery disease (CAD). However, 5% to 15% of all MI cases present with myocardial infarction with non-obstructed coronary arteries (MINOCA), a condition characterized by clinical signs of myocardial infarction but without significant stenosis (>50%) in the epicardial coronary arteries upon angiography 1. Unlike myocardial infarction with obstructive coronary artery disease (MICAD), the primary reason for which is atherosclerosis and consequent plaque rupture leading to intravascular thrombosis and obstruction, MINOCA is characterized by the absence of significant coronary obstruction, with a heterogeneous pathophysiology, which may include plaque disruption, coronary microvascular dysfunction, coronary vasospasm, and thrombosis.2 Increasing evidence suggests that inflammation plays a central role in both MINOCA and MICAD, potentially through different mechanisms. In MICAD, inflammation is well established as a key driver of atherosclerotic progression, plaque destabilization, and thrombosis, with elevated inflammatory biomarkers such as C-reactive protein (CRP), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α) correlating with worse cardiovascular outcomes 3, 4. In contrast, MINOCA is increasingly recognized as an inflammatory condition, with studies demonstrating elevated inflammatory markers in affected patients 5, 6. Moreover, imaging techniques such as pericoronary fat attenuation index and FDG-PET have provided further evidence of heightened coronary inflammation in MINOCA 7. Given the prognostic implications of inflammation in both conditions, a deeper understanding of its role in MINOCA compared to MICAD may improve diagnostic accuracy, risk stratification, and therapeutic strategies. This review aims to explore the pathways and mechanisms of inflammation in MINOCA, draw comparisons with MICAD, and highlight emerging biomarkers and imaging modalities that may aid in clinical decision-making.
Inflammation is now widely recognized as a central contributor to the pathogenesis of MICAD. Over the past decades, researchers have explored the roles of both systemic and local inflammatory processes, employing biochemical assays, advanced imaging modalities, and interventional studies to elucidate the role of inflammation, showing its duality by its participation in plaque formation and destabilization, contributing to atherothrombosis, but also influencing myocardial healing post-infarction.7, 8
At the molecular level, the inflammatory process is often initiated by endothelial dysfunction. Risk factors such as hyperlipidemia, hypertension, and smoking lead to endothelial injury, which leads to vascular permeability that allows low-density lipoprotein (LDL) to get into the intima. Subsequent oxidative modification of LDL particles triggers an inflammatory response by activating resident macrophages, which release cytokines such as IL-6 and TNF-α. These cytokines activate transcription factors such as nuclear factor-kappa B (NF-κB), resulting in the upregulation of multiple pro-inflammatory genes.9
Immune cell recruitment is another critical component of the inflammatory process in MICAD. Monocytes migrate to the area of endothelial injury and, upon entering the intima, differentiate into macrophages that engulf oxidized LDL to form foam cells and secrete additional cytokines and matrix metalloproteinases that break down the fibrous cap of atherosclerotic plaques, increasing their vulnerability 10, 11. Neutrophils also contribute by releasing reactive oxygen species (ROS), which amplify oxidative stress and further damage the vascular endothelium, reinforcing the inflammatory response 12.
Another key molecule that has recently gained attention is the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome which plays a critical role in the inflammatory processes underlying MICAD. As a cytosolic sensor, NLRP3 detects various stress signals, including oxidative stress, cholesterol crystals, and mitochondrial dysfunction, which are common in atherosclerosis and ischemic heart disease 13. Activated NLRP3 triggers a cascade of inflammatory responses that contribute to plaque instability, myocardial injury, and adverse post-infarction remodeling by combining with apoptosis-associated speck-like protein (ASC) and caspase-1, leading to the cleavage and maturating of the pro-inflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18). These cytokines amplify the local inflammatory response, exacerbating endothelial dysfunction and accelerating plaque destabilization 14. In macrophages within atherosclerotic plaques, NLRP3 activation also promotes foam cell formation and necrotic core expansion, two key contributors to plaque rupture and acute coronary events 15. Beyond its role in plaque instability, the NLRP3 inflammasome is actively involved in post-MI myocardial remodeling. Its sustained activation following myocardial infarction leads to excessive fibrosis, cardiomyocyte apoptosis, and impaired cardiac repair, which contribute to ventricular dysfunction and an increased risk of heart failure 15.
All these mechanisms make NLRP3 inflammasome an intriguing factor in “acute on chronic” inflammation in MICAD. This concept reflects the interplay between long-standing low-grade inflammation and the sudden inflammatory rise during an acute ischemic event. Patients with chronic CAD often exhibit persistent low-level inflammation, characterized by CRP, IL-6, and TNF-α, which contribute to plaque progression and vulnerability 12, 16. However, during an acute MI, an increase in inflammatory response occurs, exacerbating myocardial injury and increasing the risk of complications such as infarct expansion, adverse ventricular remodeling, and heart failure 3. Chronic NLRP3 activation in macrophages in atherosclerotic plaques promotes low-grade inflammation and plaque instability. Upon plaque rupture, NLRP3 is further activated, triggering the release of IL-1β and IL-18, which intensify myocardial damage and recruit neutrophils to the infarcted area 17. The sudden inflammatory surge results in a paradoxical effect: while inflammation is necessary for tissue repair, excessive or prolonged inflammation can worsen ischemic injury and lead to fibrosis and heart failure 3.
Targeting the NLRP3 inflammasome has been proposed as a therapeutic approach to mitigate both chronic and acute inflammatory responses in myocardial damage and to reduce post-MI complications, positioning it as a promising therapeutic target in MICAD18. Inhibitors like MCC950 and Dapansutrile have been shown to reduce infarct size and preserve cardiac function 19, 20. These findings suggest that modulating the inflammatory response—particularly during the transition from chronic to acute inflammation—may improve post-MI outcomes and prevent adverse remodeling 18.
The clinical relevance of inflammation in MICAD extends beyond its mechanical role. Several studies have examined systemic inflammatory markers, such as CRP, IL-6, and TNF-α, and their correlation with disease severity and outcomes in MICAD patients. Elevated IL-6 levels have been consistently linked to a higher risk of recurrent cardiovascular events, suggesting that systemic inflammation is not merely a consequence but an active driver of plaque progression and myocardial damage resulting in residual risk. 16, 21
A number of studies demonstrated that hs-CRP levels were predictive of recurrent ischemic events even after adjusting for traditional risk factors, putting into focus the clinical value of inflammatory biomarkers in risk stratification 22. This aligns with findings from the aforementioned studies 16, 21 that identified IL-6 and TNF-α as independent predictors of infarct size and left ventricular dysfunction post-MI. When comparing these findings, it is evident that hs-CRP provides an accessible measure of systemic inflammation, while IL-6 and TNF-α offer deeper insights into active inflammatory processes contributing to myocardial injury. These biomarkers could be integrated into clinical risk models to improve MICAD prognosis and guide treatment strategies.
Interestingly, in the CANTOS trial, a study that used IL-1β inhibitor canakinumab, results showed that reducing inflammation without altering lipid levels significantly lowered the incidence of recurrent myocardial infarction 8. This landmark study put into clinical perspective that targeting inflammation directly could yield clinical benefits, paving the way for novel anti-inflammatory interventions in MICAD.
While systemic markers reflect generalized inflammation, studies have increasingly focused on localized coronary inflammation which is increasingly recognized as a more precise indicator of plaque instability and cardiovascular risk. Advances in imaging techniques have facilitated the non-invasive assessment of coronary artery inflammation, particularly through pericoronary fat attenuation index (FAI) and FDG-PET imaging.
One study that used coronary computed tomography angiography (CCTA) with pericoronary FAI analysis demonstrated that an increased pericoronary fat attenuation, especially above s –70·1 Hounsfield units (HU), localized in the proximal segment of the right coronary artery or on the left descending artery correlated with plaque vulnerability and increased risk of major adverse cardiovascular events (MACE)23. These findings are consistent with another imaging-based study that identified higher coronary artery inflammation using FDG-PET in patients’ acute settings compared to more stable CAD patients 24.
A systemic review published in 2022 by Sagris et al. suggests that pericoronary inflammation may provide additional prognostic value beyond traditional risk factors and systemic inflammatory markers 25. Moreover, the same meta-analysis has highlighted the heterogeneous distribution of inflammation in MICAD patients, with some plaques exhibiting high inflammatory activity, thus making them more hemodynamically significant despite minimal luminal stenosis with every increase in the value of FAI 25. These findings challenge the traditional view that plaque burden alone dictates ischemic risk, emphasizing the importance of plaque composition and inflammatory status. While blood-derived biomarkers offer systemic insights into inflammation, CCTA-based fat attenuation provides direct visualization of localized coronary inflammation, making it a promising tool for early risk stratification in MICAD.
The “acute on chronic” inflammation model has gained attention as a framework for understanding the interplay between pre-existing low-grade inflammation and the acute inflammatory surge during MI. Chronic CAD patients often exhibit persistent low-level inflammation, which serves as a pro-inflammatory baseline that primes the vascular environment for an exaggerated immune response following plaque rupture 3, 26.
LATITUDE-TIMI 60 study found that patients with elevated IL-6 and TNF-α levels at four weeks post-randomization experienced worse outcomes following an acute MI, likely due to enhanced leukocyte infiltration and prolonged inflammatory activity 21. This is further supported by findings demonstrating that monocyte activation and neutrophil infiltration that are mainly driven by the chemokine MCP-1 were significantly higher in MICAD patients compared to those with stable angina 27, 28.
Comparing these findings, it is evident that chronic inflammation not only predisposes plaques to rupture but also exacerbates myocardial injury once an infarction occurs. This dual-phase inflammatory response may explain why MICAD patients with high baseline inflammation often experience larger infarcts and worse ventricular remodeling post-MI 26.
Given the well-established role of inflammation in MICAD, several studies have tried to move the research in the direction of finding anti-inflammatory therapies to improve patient outcomes. Statins, colchicine, and IL-1β inhibitors have shown promising results in modulating the inflammatory response, but their clinical efficacy varies depending on the phase of inflammation (chronic vs. acute) 17.
The Colchicine Cardiovascular Outcomes Trial (COLCOT) found that administering low-dose colchicine significantly reduced cardiovascular events in MICAD patients 29. Similar results are being detailed in another study, and also showed that 0.5mg of colchicine daily to patients with chronic CAD correlates with a decreased risk for MACE events. 30
The CANTOS study, which analyzed the effects of IL-1β inhibition via canakinumab, had promising results, further supporting inflammation as a therapeutic target. In this study patients showed a decrease in risk for MACE events by 15% and also a 24% lower risk of a recurring MI. The risk for cardiovascular death was lowered by 10% when administering canakinumab in patients post-MI. 8
Another anti-inflammatory therapy that has been studied in the context of post-MI patients is anakinra. One of the main findings on the effect of anakinra in this population was described in a pooled analysis of the VCUART clinical trials; these showed that patients that had received the treatment for 14 days post-STEMI had a reduced risk of heart failure at 1 year compared to the placebo group. 31 Behind the clinical findings it can also be seen that anakinra leads to lower levels of CRP 31 and a lower leukocyte count 32, which demonstrates the critical role the inflammation process plays in the prognostic of these patients.
Moreover, while statins exhibit anti-inflammatory effects beyond lipid-lowering, their impact on plaque-specific inflammation remains limited 33, reinforcing the need for targeted therapies such as NLRP3 inhibitors. One of the molecules studied in NLRP3 inhibition is MCC950. One study done on mice that had induced MI by ligaturing the left descending artery showed that the administration of MCC950 for 14 days decreased the amount of fibrosis post-MI and alleviated cardiac function.34
However, challenges remain. Some studies have noted that while reducing inflammation improves outcomes, excessive suppression may impair myocardial healing, suggesting a need for personalized anti-inflammatory strategies 35.
MINOCA is a distinct clinical entity characterized by myocardial infarction (MI) in the absence of significant coronary artery obstruction (>50% stenosis) on angiography, underlying that behind the clinical manifestations of a myocardial infarction lies a wide spectrum of mechanisms, far beyond the well-known atherothrombotic process 1. Despite the clinical similarities between MINOCA and MI-CAD, such as ECG findings (Fig. 1, Fig. 2), elevated cardiac enzymes and symptoms, MINOCA has a diverse pathophysiological spectrum, including coronary vasospasm, thromboembolism, coronary microvascular dysfunction (CMD), spontaneous coronary artery dissection (SCAD), and myocardial disorders such as myocarditis and takotsubo cardiomyopathy 36, which angiographically present as non-obstructed coronary arteries compared to the obstruction seen in MI-CAD (Fig. 1, Fig. 2).
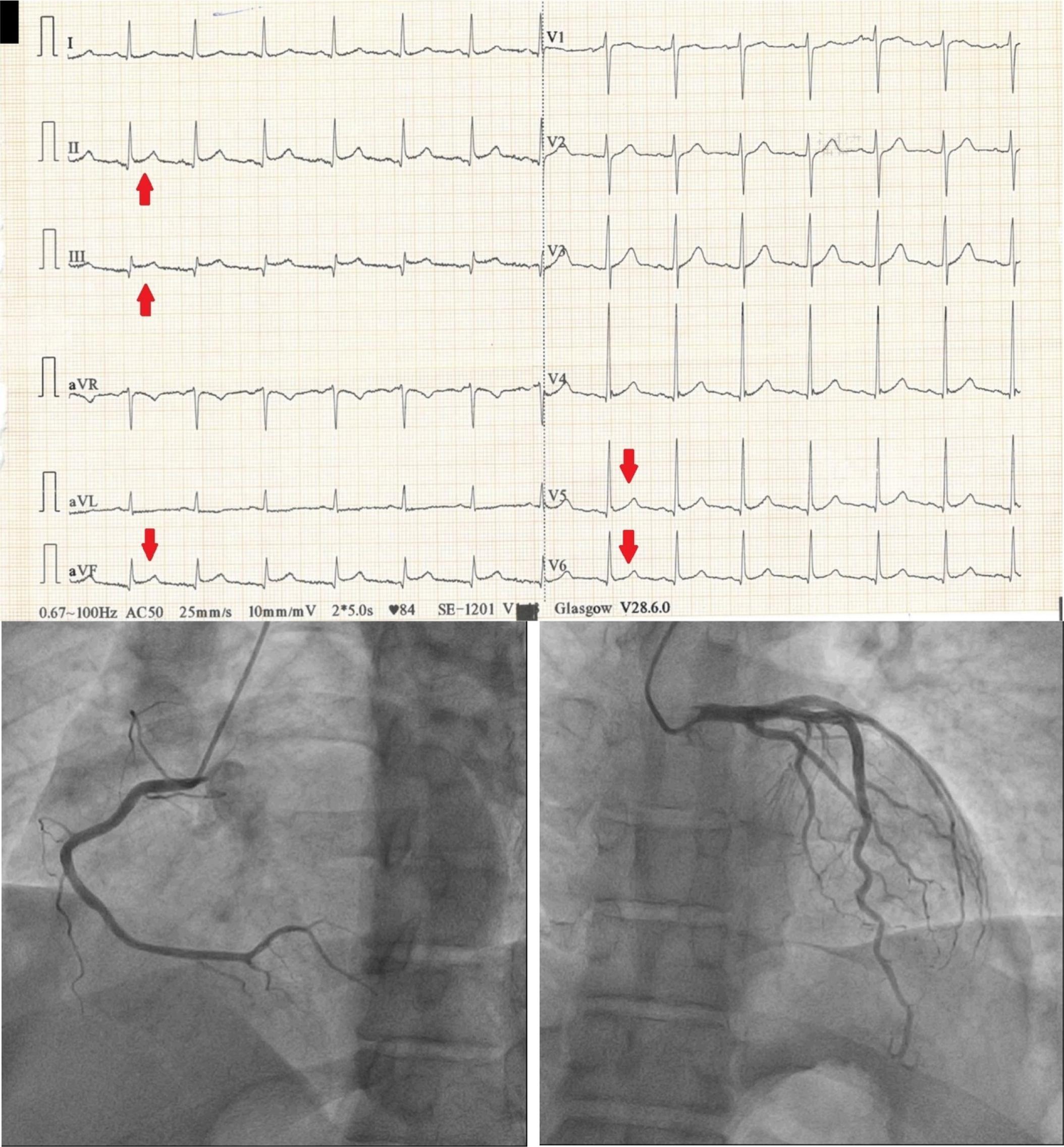
38- year old patient presenting to the E.R. with recent onset chest pain. ECG shows ST elevation in DII, DIII, aVF V5 and V6 (red arrows), suggesting a inferolateral STEMI. Patient undergoes coronary angiogram which reveals normal coronary arteries with no significant stenosis.
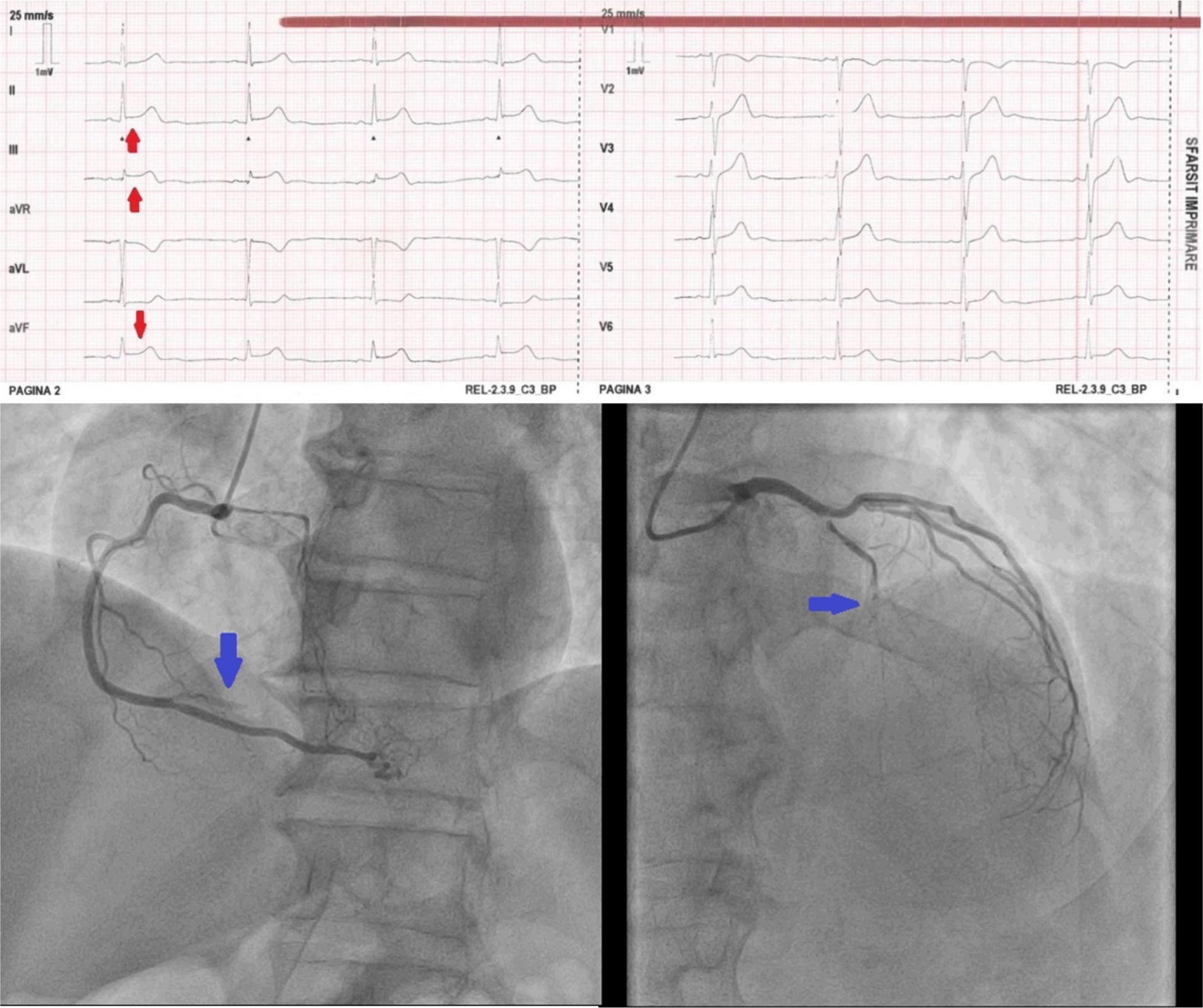
63-year old patient presenting with a day old chest pain. ECG shows ST elevation in inferior leads DII, DIII, aVF (red arrows) consistent with an inferior STEMI. Angiogram shows acute occlussion on the right coronary artery (blue arrow – left image) and chronic occlussion of the left anterior descending coronary artery (blue arrow – right image).
The heterogeneity of MINOCA makes it difficult to manage using standard MI protocols. Unlike ST-elevation myocardial infarction (STEMI) or non-ST-elevation myocardial infarction (NSTEMI) due to atherosclerotic occlusion MINOCA may not respond to conventional antiplatelet therapy in the same way 37. The growing understanding of imaging, biomarker analysis, and pathophysiological research has improved our approach to diagnosing and managing this condition.
Coronary vasospasm is a significant mechanism underlying MINOCA, leading to transient myocardial ischemia due to reversible vasoconstriction of coronary arteries. Unlike atherosclerotic obstructions, vasospasm results in dynamic narrowing that can be triggered by endothelial dysfunction, oxidative stress, or autonomic nervous system dysregulation. Coronary vasospasm is categorized into epicardial vasospasm, affecting large coronary arteries, and microvascular dysfunction, involving the smaller coronary arterioles that regulate myocardial perfusion 38.
Epicardial vasospasm is characterized by a temporary, reversible constriction of major coronary arteries, which can lead to myocardial ischemia, angina, and even infarction. This condition is frequently associated with endothelial dysfunction, as the nitric oxide-dependent vasodilatory response is impaired, resulting in an exaggerated vasoconstrictive reaction to stimuli such as acetylcholine, cold exposure, and emotional stress 39. Studies have demonstrated that patients with MINOCA often exhibit a heightened vasospastic response to acetylcholine provocation testing, supporting the role of endothelial dysfunction in its pathogenesis 40.
A key contributor to endothelial dysfunction in MINOCA is oxidative stress, which disrupts nitric oxide synthesis and bioavailability, thereby impairing vasodilation. Hung et al. highlighted that vascular inflammation, oxidative stress, and autonomic imbalance play a crucial role in increasing coronary vasoreactivity, predisposing individuals to vasospasm 41.
Beyond epicardial involvement, CMD is also a major contributor to vasospastic MINOCA. CMD results in impaired vasomotor control, abnormal coronary flow regulation, and increased microvascular resistance, leading to inadequate perfusion despite angiographically normal epicardial arteries. Recent studies suggest that CMD is one of the predominant mechanisms of MINOCA, with up to 30% to 50% of cases involving some form of microvascular dysfunction 42. The dysfunction in CMD is often related to endothelial-independent mechanisms, such as smooth muscle hypercontractility and heightened sympathetic nervous system activity 38. CMD may coexist with epicardial vasospasm, compounding ischemic risk and making diagnosis more complex.
Coronary thromboembolism and plaque disruption are other important mechanisms in MINOCA, and was found in around 30% to 40% of patients with MINOCA 43. These conditions result in myocardial ischemia due to either embolization of thrombotic material into the coronary circulation or disruption of a vulnerable plaque that does not lead to complete luminal occlusion. The pathophysiology of these mechanisms is diverse, involving factors such as thrombophilia, atrial fibrillation (AF), left ventricular thrombi, paradoxical embolism, and minor plaque erosion.
Coronary thromboembolism refers to the embolization of thrombotic material into the coronary arteries, which may originate from cardiac or extracardiac sources. Cardiac sources include atrial fibrillation, left ventricular thrombi, infective endocarditis, and prosthetic heart valves, while extracardiac sources involve paradoxical embolism via a patent foramen ovale (PFO) or aortic atheromas 44.
Opolski et al. demonstrated that some MINOCA cases, even if they were less than MICAD cases, involve embolic occlusions without evidence of coronary atherosclerosis 45, emphasizing the need for thrombophilia screening in high-risk patients. Other studies looked further into the correlation between thrombophillic diseases and their connection to MINOCA, and found that patients who suffered from such an event had higher rates of thrombophilia. 46, 47 Additionally, Reynolds et al. used OCT and CMR to identify thromboembolic sources in MINOCA, finding that a significant subset of patients had embolic infarcts 43.
Because coronary embolism can be transient, angiographic imaging may fail to detect evidence of an obstruction. This highlights the importance of multimodal imaging, including transesophageal echocardiography (TEE) and contrast-enhanced cardiac MRI, to detect embolic sources that may not be evident with standard coronary angiography 42.
Unlike coronary embolism, plaque disruption in MINOCA occurs when an atherosclerotic plaque erodes or ruptures without leading to total occlusion. This mechanism is more commonly associated with younger patients and women, who often present with MINOCA without significant obstructive disease. Plaque disruption is typically classified into two main forms: plaque rupture characterized by a fibrous cap rupture that leads to exposure of lipid-rich plaque contents, triggering thrombosis and ischemia, and plaque erosion in which the endothelial surface becomes disrupted without rupture, leading to localized thrombus formation 48, 49. The ability to detect subtle plaque disruption using these imaging techniques highlights the importance of intracoronary imaging in MINOCA evaluation.
SCAD is an important but underrecognized cause of MINOCA. It primarily affects younger women and is associated with hormonal fluctuations, fibromuscular dysplasia, and extreme emotional or physical stress. Unlike atherosclerotic coronary artery disease, SCAD occurs due to spontaneous tearing of the arterial wall, leading to an intramural hematoma that compresses the lumen and results in myocardial ischemia 50. This phenomenon has been seen in patients who also had associated systemic inflammatory diseases or connective tissue disorders49.
SCAD is often misdiagnosed due to its transient and dynamic nature, with angiographic findings sometimes appearing normal or inconclusive. CMR imaging and intracoronary imaging techniques such as OCT and IVUS are essential for diagnosing SCAD in MINOCA patients 42. The use of OCT has been highlighted as particularly effective in identifying wall abnormalities and distinguishing SCAD from other causes of MINOCA, such as myocarditis or takotsubo cardiomyopathy 51.
SCAD-related MINOCA differs from other MINOCA etiologies in terms of management. Unlike classic acute coronary syndrome (ACS), the use of anticoagulation, thrombolytics, and aggressive percutaneous coronary intervention (PCI) can be detrimental and may worsen the dissection. Unless the patient presents with hemodynamic instability, a conservative approach is often preferred, with beta-blockers, lifestyle modifications, and close monitoring being the mainstay of treatment 52.
MINOCA is a complex cardiovascular condition involving diverse pathophysiological mechanisms. Among these, inflammation plays a crucial role in its onset and progression. Inflammatory molecules and pathways have been increasingly implicated in MINOCA, from the shared mechanisms with MICAD to finding specific key factors in MINOCA. Understanding the involvement of key inflammatory markers, cytokines, and pathways is essential for elucidating the underlying mechanisms and improving diagnostic strategies.
Several inflammatory markers that were found to play a role in MICAD have been also linked to MINOCA, including hs-CRP, interleukins, and TNF-α. Elevated levels of hs-CRP are frequently found in MINOCA patients, suggesting a systemic inflammatory state 53.
A study identified IL-6 as a crucial mediator of vascular inflammation, which plays a role in endothelial dysfunction and microvascular dysfunction, both key features of MINOCA. IL-6 has been associated with increased coronary microvascular resistance and impaired vasodilation, contributing to ischemia even in the absence of epicardial obstruction 54. Furthermore, hs-CRP, a well-established marker of systemic inflammation, is significantly elevated in MINOCA patients, indicating ongoing vascular and myocardial inflammation. Elevated hs-CRP levels are associated with increased cardiovascular risk and worse clinical outcomes 5.
Endothelial dysfunction is a hallmark of MINOCA, often driven by pro-inflammatory cytokines. Studies have demonstrated that TNF-α induces endothelial damage, reduces nitric oxide bioavailability, and promotes vascular inflammation 53. These effects contribute to CMD, a primary mechanism in MINOCA.
Another crucial inflammatory pathway involved in MINOCA is the IL-1 signaling cascade. IL-1β has been shown to promote endothelial activation, leading to increased expression of adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), which facilitate leukocyte infiltration and perpetuate vascular inflammation 55. Increased IL-1 activity has also been linked to myocardial fibrosis, suggesting that chronic inflammatory activation contributes to long-term structural changes in the myocardium 56.
In addition to IL-1, IL-18, a member of the IL-1 superfamily, has been shown to amplify the inflammatory response by promoting monocyte and macrophage activation, which enhances the release of additional pro-inflammatory cytokines and increases oxidative stress. These inflammatory cascades collectively lead to a sustained pro-thrombotic environment, increased endothelial permeability, and microvascular dysfunction, all of which are crucial mechanisms in MINOCA pathogenesis 57.
A newer published review 58 highlights the role of the cytokine trio – visfatin, placental growth factor (PlGF), and fractalkine in MINOCA-related inflammation and vascular dysfunction. Visfatin, secreted by adipose tissue and immune cells, acts as a pro-inflammatory mediator by activating NF-κB, increasing cytokine production, and promoting endothelial dysfunction. PlGF, part of the vascular endothelial growth factor (VEGF) family, enhances monocyte infiltration and neovascularization but also induces oxidative stress and plaque destabilization. Fractalkine, a chemokine, binds to CX3CR1 receptors on monocytes and T cells, amplifying vascular inflammation and endothelial activation.
These cytokines collectively worsen microvascular dysfunction and inflammatory injury, making them potential therapeutic targets. Visfatin amplifies PlGF and fractalkine signaling: by activating NF-κB, visfatin upregulates the expression of VEGFR-1 and CX3CR1, increasing the sensitivity of endothelial cells to PlGF and fractalkine-induced inflammation. This synergy enhances monocyte adhesion and infiltration into the vessel wall, worsening vascular inflammation. PlGF-induced oxidative stress reinforces fractalkine activation: PlGF-mediated ROS production exacerbates endothelial dysfunction, leading to increased shedding of fractalkine from endothelial cells. This results in heightened monocyte and macrophage recruitment, fueling a persistent inflammatory state. Fractalkine sustains visfatin and PlGF-driven inflammation by maintaining leukocyte adhesion and activation, fractalkine ensures sustained production of visfatin and PlGF, creating a chronic inflammatory milieu that contributes to microvascular instability, endothelial damage, and impaired coronary perfusion. 58
Inflammation and oxidative stress are closely intertwined in MINOCA. Oxidative stress exacerbates endothelial dysfunction by increasing ROS, impairing nitric oxide production, and enhancing vascular inflammation 58. Elevated ROS levels have been linked to platelet hyperactivation, endothelial apoptosis, and increased thrombotic risk, all of which may contribute to the ischemic events seen in MINOCA 14.
Moreover, immune cell infiltration into coronary microcirculation has been observed in MINOCA patients. Macrophages and neutrophils, key players in the immune response, contribute to vascular inflammation and endothelial disruption. A study using CMR imaging showed that patients with MINOCA had increased myocardial inflammation, supporting the role of immune activation in the disease process. Additionally, activated monocytes and neutrophils secrete matrix metalloproteinases (MMPs), which degrade the extracellular matrix and contribute to microvascular instability 14.
As discussed in the MICAD mechanisms, chronic low-grade inflammation is a major contributor to CMD 59 which is one of the leading causes of MINOCA. Inflammatory cytokines and oxidative stress induce microvascular constriction, capillary rarefaction, and impaired coronary flow reserve, leading to ischemia even in the absence of obstructive coronary artery disease 56. CCTA studies have confirmed that patients who do not present obstructive CAD often exhibit microvascular inflammation and endothelial swelling, further supporting the role of chronic inflammation in disease progression 60.
Additionally, autoimmune conditions, including systemic lupus erythematosus and rheumatoid arthritis, have been associated with an increased prevalence of MINOCA. Autoimmune-mediated vasculitis and endothelial injury may contribute to coronary microvascular dysfunction and ischemia in these patients 49. This further raises the question of whether elevated levels of circulating immune complexes and autoantibodies suggest an autoimmune-driven component in MINOCA pathogenesis, highlighting the need for investigation into the immune mechanisms of MINOCA.
All the information presented earlier lead to the fact that inflammation plays a pivotal role in the pathogenesis of MINOCA, and the identification of inflammatory pathways involved in MINOCA not only enhances our understanding of its mechanisms but also paves the way for improved diagnostic approaches. Future research should focus on biomarker-guided personalized treatment strategies to improve outcomes in MINOCA patients.
As stated before, MINOCA is a complex and heterogeneous condition that accounts for approximately 6% to 15% of all MI cases. Unlike obstructive CAD, MINOCA lacks significant stenotic lesions on angiography, making prognosis assessment and management strategies particularly challenging. While previously considered a benign condition, recent studies have shown that MINOCA patients face substantial risks of recurrent cardiovascular events, heart failure, and mortality 61. We aim to do an analysis of the prognosis of MINOCA based on the findings of multiple studies, emphasizing mortality rates, recurrence risks, biomolecular markers, and the impact of inflammatory and metabolic factors.
Several studies have evaluated the long-term survival of MINOCA patients, with mortality rates varying significantly depending on underlying pathophysiological mechanisms and comorbidities. According to a meta-analysis published in 2022 that look at 44 articles which comprised 36,932 showed that the mortality rate was 2.0% 62. Another later study published in European Heart Journal found that the 5-year mortality rate was approximately 8.5%, with higher mortality among elderly individuals and those with systemic inflammatory disorders 6, and the mortality rate went up to 16% at three-year follow-up in a study by Zhang et al 61.
Sex differences in MINOCA outcomes have also been noted. In one study of MINOCA cases women were represented more, with 168 (65.9%) vs. 87 male patients, while in another larger retrospective study women made up 62% of all cases recorded 63. But as far as the mortality rate between the two sexes, multiple studies didn’t find any significant differences between the two groups 64, 65. Additionally, age has emerged as a significant predictor of mortality, with individuals over 70 years old having a higher mortality rate than younger patients, especially in female patients who had a mortality rate of 20.7% compared to 2.6% for those under 70 years. 64
Further research has suggested that the presence of systemic inflammatory diseases, such as rheumatoid arthritis and systemic lupus erythematosus, may contribute to the onset of MINOCA, but it doesn’t correlate with an increase in mortality for these patients after a median follow-up period of 28 months. One study reported that MINOCA patients had a higher incidence of pro-inflammatory conditionds, but the occurrence of MACE or mortality didn’t correlate with the these conditions. 59
As stated before, MINOCA is not a benign condition. The risk of major adverse cardiovascular events (MACE) has been studied in MINOCA–only cohorts as well as in comparison to MI-CAD. A study published in International Journal of Cardiology that included 2147 patients with MINOCA found that at 4.5 years, 24% of the patients have experienced MACE. 63 Out of all MACE, deaths represented most of the events, with a total of 1,254, of which 1,165 happened after the first 30 days and only 497 (42.7%) had a cardiovascular cause. 63
The aforementioned study by Chaudhary et al. has shown that even though the mortality rate doesn’t differ between genders, the MACE risk is higher in women: 10.1% compared to 9.1%, and even more women displayed higher rates of recurrent ischemic events: 3.5% vs. 2.2%, indicating potential disparities in disease mechanisms and responses to treatment 65. These findings are supported by another study that also showed that the incidence of MACE in women, especially under 70 years of age, was higher than in men. 64 Even though this study has not seen any differences in MACE when analyzing it by age, there are more studies published that show age as an independent factor in the appearance of MACE in MINOCA patients. A large retrospective study that looked at 1,544 patients over a period of seven years has found that age does correlate with the risk of MACE, but the end point was only comprised of all-cause mortality and MI recurrence. 66 In support of these findings comes the Nordenskjöld et al. study that compared 10-year-difference age groups, and showed that the risk of MACE increased alongside the increase in age. 63
Another traditional risk factor that is looked at in most of the studies that followed the outcomes of MINOCA patients is the lipid profile. And even though dyslipidemia has been less observed in MINOCA patients, it is still an important factor in the prognosis. In one study LDL-C has been linked to a higher risk of MACE 67, and in another study is was shown that residual cholesterol is a factor that contributes to an adverse prognosis for patients with MINOCA 68. A newer direction for study is the effect of lipoprotein (a) [Lp(a)] in MINOCA, given its known effect on poor outcome in MICAD patients. One study focusing on the effect of Lp(a) analyzed the risk of MACE appearence in 1,179 patients with MINOCA and showed that higher levels of Lp(a) defined as over 30mg/dL, correlated with a higher rate of MACE over a median follow-up period of 41.7 months. Even more, it described an increase in risk paralleled with the increase in Lp(a) levels. 69 In addition to this, a newer study published in 2023 showed that beside the already mentioned risk factors such as age, a Lp(a) level higher than 60mg/dL was considered an independent risk factor for worse outcomes in MINOCA patients. 70
Biomarkers in MINOCA.
| Biomarkers | Mechanisms | Changes in MINOCA | Effect on prognosis |
|---|---|---|---|
| C-reactive protein (CRP) 5 6, 73 | Vascular and myocardial inflammation | In MINOCA vs. MI-CAD | Elevated risk of mortality and MACE |
| Tumor necrosis factor α (TNF-α) 53 |
| In MINOCA vs MI-CAD | Unknown |
| Interleukin-6 (IL-6) 21, 54 |
| In CMVD | Elevated risk of recurrent myocardial infarction |
| Interleukin-1 (IL-1) 55, 56 |
| In MINOCA | Unknown |
| Visfatin 58 |
| In MINOCA | Unknown |
| Placental growth factor (PIGF) 58 |
| In MINOCA | Unknown |
| Fractalkine 58 |
| In MINOCA | Unknown |
| Fibrinogen-on-albumin ratio (FAR) 74 |
| - | Higher risk of MACE and hospitalization for heart failure |
| sVCAM-1 55 |
| In MINOCA vs MI-CAD | Progression of atherosclerosis |
| Soluble urokinase-type plasminogen activator receptor (suPAR) 80 |
| In MINOCA vs. healthy control | Increase in MACE and all-cause deaths. |
| Uric acid 79 |
| - | Higher risk of MACE and heart failure |
| Lipoprotein(a) 69, 70 |
| In MINOCA vs MI-CAD | Increase in MACE |
| Ratio of neutrophils, monocytes, lymphocytes, platelet to HDL (NHR, MHR, LHR, PHR) 75 |
| Increase in MACE | |
| Systemic inflammation response index (SIRI) 76 | - | - | Higher rates of MACE |
| Neutrophil-to-lymphocyte ratio (NLR) 77 |
| In MINOCA | Increased risk of all-cause mortality |
| White blood cell count to mean platelet count ratio (WMR) 78 |
| - | Higher rates of MACE |
| Neutrophil-to-platelet ratio 78 |
| - | Higher rates of MACE |
Other traditional risk factors seen to play a factor in MI-CAD prognosis, such as diabetes, chronic kidney disease, and a low left ventricular ejection fraction (LVEF) <40%, were also found to impact the outcome of patients that suffered a MINOCA, by increasing the risk of MACE as well as the risk of mortality. 71
Inflammation plays a critical role in the prognosis of MINOCA, influencing both acute and long-term cardiovascular outcomes. Unlike obstructive myocardial infarction, MINOCA is characterized by coronary microvascular dysfunction, endothelial inflammation, and pro-thrombotic activity, which contribute to persistent ischemia, recurrent events, and increased mortality risk. Numerous studies have demonstrated that inflammatory markers, including CRP, IL-6, fibrinogen, and oxidative stress-related molecules, serve as significant predictors of prognosis in MINOCA patients.
CRP levels have been one of the first to be studied when looking into inflammation and the effect it has on the outcomes of patients with MINOCA. A Journal of the American Heart Association (JAHA) study highlighted that CRP levels decreased faster over a month in MINOCA patients than in MI-CAD patients, correlating with a lower extent of the MI. 72 A post-hoc of the PLATO study has found that higher levels of CRP were an independent factor that predicted all-cause mortality, and it also correlated with a higher risk for MACE 73, results that are also seen in a retrospective study from European Heart Journal which looks at 706 patients over a median of five years and has showed that the CRP at admission was an independent factor for all-cause mortality. 6 Elevated IL-6 levels, a key pro-inflammatory cytokine, have also been linked to a an increased risk of recurrent myocardial infarction, emphasizing the contribution of endothelial injury and immune activation to long-term cardiovascular complications21. Another study reported that patients with persistent inflammation had a significantly higher risk of long-term cardiovascular mortality, suggesting that chronic systemic inflammation plays a crucial role in the pathophysiology of MINOCA and its long-term outcomes. 5
The impact of fibrinogen, an acute-phase reactant involved in coagulation, has also been well established. One study published in the International Journal of Cardiology looked at fibrinogen-on-albumin ratio (FAR) and reported that patients with MINOCA who had high FAR levels demonstrated a higher risk of major MACE, with a 22% rate of occurrence compared to 9.3%, and also greater rate of hospitalization for heart failure. 74
A growing body of research has also identified oxidative stress and endothelial dysfunction as key contributors to poor prognosis in MINOCA. In one study the progression of atherosclerosis over a period of one year in MINOCA patients has been closely linked to sVCAM-1 and CCL-21, indicating a multifaceted underlying mechanism. These markers suggest that atherosclerosis progression may be driven by a combination of structural plaque alterations and microcirculatory dysfunction, highlighting the complex interplay between vascular inflammation and endothelial integrity. 55 Further supporting these findings, asymmetric dimethylarginine (ADMA), an endogenous inhibitor of nitric oxide synthesis, was significantly elevated in MINOCA patients, correlating with endothelial dysfunction and increased long-term cardiovascular risk 53.
A study published in Frontiers in Cardiovascular Medicine looked at the role of multiple inflammatory biomarkers: ratio of neutrophil count to HDL level (NHR), ratio of monocyte count to HDL level (MHR), ratio of lymphocyte count to HDL level (LHR), ratio of platelet count to HDL level (PHR), systemic immune-inflammation index (SII), systemic inflammation response index (SIRI), and aggregate index of systemic inflammation (AISI).The results demonstrated that the MACE group had increased oxidative stress markers and the association of NHR, MHR, PHR, SII, SIRI, and AISI are a good predictor for the recurrence of MACE, reinforcing the role of inflammation in MINOCA. 75 SIRI biomarker has also been researched in correlation with the role of N-terminal Pro-B-type natriuretic peptide (NT-proBNP) and showed that high levels of SIRI correlate strongly with worse outcomes and higher rates of MACE, and can be a good indicator for risk, especially when combined with NT-proBNP levels. 76
Another significant biomarker that has been shown to have increased levels and also play a role in the prognosis of patients with MINOCA is neutrophil-to-lymphocyte ratio (NLR). In one study that analyzed MINOCA patients versus control patients that had normal angiographic coronary arteries, patients presenting with MINOCA had significantly higher levels of NLR and that these levels correlated with an increase in mortality over a median period of 21 months. 77 These results correlate with the findings of another study that has investigated the impact of white blood cell count to mean platelet volume ratio (WMR) and neutrophil-to-platelet ratio (NPR). It followed 274 patients with MINOCA that were classified considering the medians of WMR and NPR. The results showed that the group with higher values of these biomarkers had higher rates of MACE, making WMR and NLR independent predictors of MACE. 78
Additionally, research published in Nutrition and Metabolism identified hyperuricemia as an independent predictor of poor prognosis in MINOCA patients, with serum uric acid levels ≥420 μmol/L in men or ≥357 μmol/L in women being associated with an increased risk of MACE of 18.7% compared to 12.8%, and also higher rates of heart failure 79. This association is thought to arise from oxidative stress, endothelial inflammation, and platelet hyperactivation, all of which contribute to recurrent cardiovascular events and impaired myocardial recovery. Similarly, the soluble urokinase□type plasminogen activator receptor (suPAR) and high-sensitivity troponin I (hsTnI) have been found to be a robust inflammatory predictor, with patients exhibiting suPAR >2523 pg/ml and a hsTnI >2.7 pg/ml demonstrating an 24 and 19-fold increased likelihood of adverse cardiovascular events as well as all-cause adverse events. 80
Given the strong correlation between inflammation and prognosis in MINOCA, there is growing interest in targeted anti-inflammatory strategies and risk stratification approaches to improve long-term outcomes. Future research should focus on developing biomarkerbased risk assessment models and personalized treatment protocols to mitigate inflammation-driven myocardial injury and recurrent ischemic events.
Advanced imaging techniques, including CMR, IVUS, and OCT, have enhanced risk stratification in MINOCA. A study published in JACC: Cardiovascular Imaging found that patients with higher percentage of late gadolinium enhancement (LGE) on CMR had an increased risk of MACE at three years. 81 Furthermore, OCT studies have detected occult atherosclerotic plaque disruptions in 52.1% of MINOCA patients, demonstrating that underlying coronary pathology is more common than initially believed. Also it has shown that the MACE risk for atherosclerotic group of MINOCA patients was higher than in the non-atherosclerotic group, especially for revascularization and rehospitalization. 82 These imaging findings correlate with higher long-term cardiovascular risk, underscoring the importance of multimodal assessment.
MINOCA and MICAD represent distinct yet overlapping clinical entities, each with unique underlying mechanisms, inflammatory profiles, and prognostic implications. Inflammation is a key driver of both MINOCA and MICAD, but its role in these conditions differs significantly. The presence of biomarkers such as sVCAM-1, CCL-21, and FAR further supports the role of endothelial inflammation and microvascular remodeling in MINOCA, distinguishing it from MICAD in terms of inflammatory pathology. Prognostically, both MINOCA and MICAD are associated with substantial long-term cardiovascular risks, though their trajectories differ. While MICAD patients have a higher short-term mortality risk due to acute coronary obstruction, MINOCA patients face increased risks of recurrent ischemic events, heart failure, and persistent microvascular dysfunction over time. Given the inflammatory underpinnings of both conditions, targeted anti-inflammatory therapies and risk stratification approaches may improve long-term outcomes. A deeper understanding of these mechanisms will enable personalized treatment strategies, reducing morbidity and mortality in both MINOCA and MICAD populations.