
Polyfunktionale Thiole zählen zu den zentralen flüchtigen Aromakomponenten in Wein und anderen fermentierten Fruchtprodukten. Sie tragen maßgeblich zum Bukett bestimmter Rebsorten bei und prägen insbesondere das aromatische Profil von Sauvignon Blanc. Zu den bedeutendsten Vertretern zählen 4-Methyl-4-sulfanylpentan-2-on (4MSP, vormals 4-MMP), 3-Sulfanylhexan-1-ol (3SH, vormals 3-MH) und 3-Sulfanylhexylacetat (3SHA, vormals 3-MHA), die mit charakteristischen Noten von schwarzer Johannisbeere, Grapefruit und Passionsfrucht assoziiert werden (Tominaga et al., 1998; Roland et al., 2011).
Diese geruchsintensiven Verbindungen liegen in den Trauben jedoch nicht in freier, aromawirksamer Form vor, sondern primär als geruchslose, gebundene Vorstufen (Precursors), insbesondere als S-Cystein-bzw. S-Glutathion-Konjugate (Peyrot des Gachons et al., 2002). Während 4MSP sowohl im Mesokarp als auch im Exokarp vorkommt, findet sich 3SH überwiegend im Exokarp von Sauvignon Blanc. Die Umwandlung dieser Konjugate in freie Thiole erfolgt enzymatisch während der alkoholischen Gärung durch β-Lyase-Aktivität. Dabei wird jedoch nur ein Bruchteil – etwa 10 % – der verfügbaren Vorstufen tatsächlich in ihre aromawirksame Form überführt (Ribéreau-Gayon et al., 2000; Swiegers et al., 2007). Diese geringe Umsetzungseffizienz unterstreicht die analytische und technologische Relevanz einer exakten Erfassung sowohl der freien als auch der potenziellen Thiol fraktionen.
Auch wenn polyfunktionale Thiole zunächst vor allem mit Sauvignon Blanc assoziiert wurden, konnten sie mittlerweile in einer Vielzahl anderer Rebsorten – sowohl in Weiß- als auch in Rotweinen – in unterschiedlich hoher Konzentration nachgewiesen werden (Rodríguez-Bencomo et al., 2009). Aufgrund ihrer niedrigen Geruchsschwellen im ng/L-Bereich und ihrer hohen sensorischen Relevanz erfordert ihre analytische Bestimmung besonders empfindliche und selektive Methoden. In den vergangenen zwei Jahrzehnten wurden verschiedene Verfahren zur Quantifizierung dieser Verbindungen entwickelt, darunter Derivatisierungstechniken gekoppelt mit Festphasenmikroextraktion (SPME) oder flüssig-flüssige Extraktionsansätze mit anschließender GC-MS-Analyse (Mateo-Vivaracho et al., 2006; Ferreira et al., 2009). Trotz dieser Fortschritte bestehen weiterhin Einschränkungen hinsichtlich der Nachweisgrenzen, Wiederholbarkeit und der Robustheit gegenüber Matrixeffekten (Wardencki, 1998; Mateo-Vivaracho et al., 2007).
In den vergangenen zwei Jahrzehnten wurden verschiedene Ansätze zur Bestimmung niedrigsiedender polyfunktionaler Thiole entwickelt. In der nachfolgenden Tabelle (Tab. 1) wird eine Übersicht der gängigsten Verfahren mit deren Nachweisgrenzen und Limitierungen angeführt. Viele dieser Methoden zeigen Einschränkungen in Bezug auf Matrixeffekte und Wiederholbarkeit, wodurch die Entwicklung einer robusteren Methode notwendig war.
Verfahren zur Bestimmung niedrigsiedender polyfunktionaler Thiole
| Studie / Plattform | Probenvorbereitung / Derivatisierung | Detektion | LOD (ng/L) |
|---|---|---|---|
| Sari et al. 2025 | SPE-Extraktion, Derivatisierung mit 1-Iodhexan und Cs2CO3, LVI-Injektion | GC-MS/MS (MRM) | 6,8 / 9,3 / 5,9 |
| Mateo-Vivaracho et al. (2007) | LLE + PFBBr-Derivatisierung | GC-NICI-MS | 0,1 / 7 / 0,6 |
| Rodríguez-Bencomo et al. (2009) | HS-SPME + PFBBr-Derivatisierung | GC-MS | < Geruchsschwelle |
| Musumeci et al. (2015) | Extractive Alkylation + HS-SPME | GC-EI-MS | 0,9 / 1 / 17 |
| Ochiai & Kishimoto (2015) | der-SBSE (ETP) | TD-GC-MS/MS | 0,20 / 27 / 0,19 |
Zielsetzung der vorliegenden Studie war die Entwicklung und Validierung einer empfindlichen und reproduzierbaren Methode zur gleichzeitigen Bestimmung der drei wichtigsten flüchtigen Thiolverbindungen (4MSP, 3SH, 3SHA) in österreichischen Weiß- und Rotweinen. Besonderes Augenmerk galt dabei der Verbesserung der Nachweisgrenzen, der Linearität und der Wiederholbarkeit der Methode unter realen Probenbedingungen. Darüber hinaus wurde untersucht, inwieweit das Thiolprofil rebsortenabhängig variiert und welche weinbaulichen oder vinifikatorischen Rückschlüsse sich daraus ableiten lassen. In einer Folgestudie ist die Erweiterung des Analysetargets um drei zusätzliche Thiolverbindungen sowie eine methodische Optimierung vorgesehen. Diese Verbindungen sind nicht nur für österreichische Weine von Bedeutung, sondern auch für aromatische Weine weltweit charakteristisch.
In Tab. 2 sind die unterschiedlichen Eigenschaften der untersuchten Thiolverbindungen ersichtlich.
Struktur, Geruchseigenschaften und -schwellenwerte der untersuchten Thiole im Wein
| Verbindung | Abkürzung | Struktur | Typische Geruchseigenschaften | Wahrnehmungsschwelle im Wein [ng/l] | Literatur |
|---|---|---|---|---|---|
| 4-Methyl-4-sulfanylpentan-2-on | 4-MSP (4-MMP) | grün-vegetabil (Buchsbaum) schwarze Johannisbeerknospe Ginster tropisch-fruchtig (Passionsfrucht) | 0,8 | Tominaga et al., 1998 Coetze et al., 2015 | |
| 3-Sulfanylhexan-1-ol | 3-SH (3-MH) | tropisch-fruchtig (Passionsfrucht, Guave) zitrusartig (Grapefruit) Stachelbeere | 60 | Tominaga et al., 1998 Coetze et al., 2015 | |
| 3-Sulfanylhexylacetat | 3-SHA (3-MHA) | intensiv tropisch-fruchtig (Passionsfrucht, Guave) zitrusartig (Grapefruit) Stachelbeere leicht grünlich (Buchsbaum) | 4,2 | Tominaga et al., 1998 Coetze et al., 2015 |
Für die Anwendung und Validierung der entwickelten Methode wurden insgesamt 160 Weinproben unterschiedlicher Rebsorten und Herkunft herangezogen. Das Probenmaterial umfasste 17 Sauvignon Blanc-Weine, 42 Grüner Veltliner Weine, 22 Gemischter Satz-Weine, 26 Blaufränkisch-Weine, 13 Zweigelt-Weine sowie 40 Schilcher-Weine. Sämtliche Proben stammten von verschiedenen Herstellern und Weinbaulagen, um eine möglichst breite repräsentative Abdeckung sortentypischer und herkunftsbedingter Unterschiede zu gewährleisten. Die detaillierten Angaben zu Rebsorte, Jahrgang und Herkunft sind in Tab. 3 zu finden.
Rebsorte, Jahrgang und Herkunft der untersuchten Weine
| Rebsorte | Anzahl | Jahrgang | Herkunft |
|---|---|---|---|
| Sauvignon Blanc | 17 | 2022 | Steiermark |
| Grüner Veltliner | 11 | 2022 | Deutschland |
| 8 | 2018 | Niederösterreich | |
| 23 | 2022 | Niederösterreich | |
| Gemischter Satz | 10 | 2022 | Wien |
| 11 | 2022 | Niederösterreich | |
| Blaufränkisch | 26 | 2022 | Burgenland |
| Zweigelt | 13 | 2021 | Niederösterreich |
| Schilcher | 40 | 2022 | Steiermark |
Die in dieser Studie verwendeten Chemikalien wiesen eine Reinheit von mindestens 95 % auf und wurden, sofern nicht anders angegeben, in analytischer Qualität eingesetzt. Die Referenzsubstanzen 4-Methyl-4-sulfanylpentan-2-on (4-Mercapto-4-methyl-2-pentanon, 98 %) und 3-Sulfanylhexan-1-ol (3-Mercapto-1-hexanol, 96 %) wurden von Thermo Scientific (Waltham, Massachusetts, USA) bezogen. 3-Sulfanylhexylacetat (3-Mercaptohexylacetat, ≥98 %) stammte von Sigma-Aldrich (St. Louis, Missouri, USA).
Weitere eingesetzte Chemikalien waren tert-Butylmethylether (≥99,5 %, Carl Roth, Karlsruhe, Deutschland), Methanol (≥99,9 %, Chem Lab, Zedelgem, Belgien), Acetonitril (≥99,95 %, Chem Lab), Ethanol (≥99,8 %, Riedel-de-Haёn, Seelze, Deutschland), Dimethylsulfoxid Rotipuran (≥99,8 %, p.a., Carl Roth), Isohexan (≥99 %, Carl Roth), sowie Natriumsulfat (≥99 %, p.a., Carl Roth).
Die Derivatisierungsreagenzien umfassten 1-Iodhexan (≥98 %, Sigma-Aldrich), Cäsiumcarbonat (≥99,9 %, p.a., Carl Roth) und Natriumborhydrid (98 %, Sigma-Aldrich). Zur Stabilisierung und Reaktionsführung wurden 4-(Hydroxymercuri)benzoesäure-Natriumsalz (≥95 %, Sigma-Aldrich), N-Acetyl-L-Cystein (≥98 %, Carl Roth), Tris(hydroxymethyl)-aminomethan (p.a., Merck KGaA, Darmstadt, Deutschland), Natriumhydroxid (Pellets, Merck), Natriumhydrogencarbonat (≥99,5 %, Carl Roth), EDTA (≥99 %, USP, Carl Roth) und Borsäure (≥99,8 %, Carl Roth) eingesetzt. Als Säurekomponente wurde L(+)-Weinsäure für Analysezwecke (≥99,5 %, Merck KGaA) verwendet.
Für die Festphasenextraktion kam Lichrolut EN (Merck KGaA) zum Einsatz.
Die folgenden Pufferlösungen wurden im Labor gemäß den analytischen Anforderungen hergestellt:
Tris-Puffer (0,1 M), mit NaOH auf pH 7,2 eingestellt
PHMB-Lösung (2 mM in 0,1 M Tris-Puffer, pH 7,2)
EDTA-Lösung (50 g/L)
Weinsäurepuffer (5 g/L), mit NaOH auf pH 3,0 eingestellt
Boratpuffer (0,2 M), mit NaOH auf pH 10,0 eingestellt
Kunstwein (4 g/L Weinsäure, 12 % EtOH), mit NaOH auf pH 3,2 eingestellt
Alle Lösungen wurden frisch zubereitet oder unter lichtgeschützten Bedingungen bei 4 °C gelagert, sofern erforderlich.
Zur quantitativen Bestimmung der Thiolverbindungen wurden isotopenmarkierte interne Standards verwendet. Dabei kamen 4MSP-d10 (≥98 %), 3SH-d5 (≥98 %) und 3SHA-d5 (≥98 %) zum Einsatz, jeweils bezogen von Eptes Sàrl (Vevey, Schweiz). Die Standards wurden in Methanol gelöst und in geeigneten Konzentrationen von je 100 ng/l als Arbeitsstandards aliquotiert und unter Stickstoff lichtgeschützt bei –20 °C gelagert. Die tatsächlichen Konzentrationen der Standards wurden wöchentlich durch Vergleichsmessungen über einen 7820A GC-FID durchgeführt (Agilent Technologies, Santa Clara, USA).
Die quantitative Bestimmung der Thiolverbindungen 4MSP, 3SH und 3SHA erfolgte mittels Gaschromatographie gekoppelt mit Tandem-Massenspektrometrie (GC-MS/MS). Die Analysen wurden auf einem 7890B-Gaschromatographen in Kombination mit einem 7010B Triple Quadrupole Massenspektrometer (beide Agilent Technologies, Santa Clara, USA) durchgeführt. Für die Probeninjektion kam ein Programmed Temperature Vaporisation (PTV)-Injektor zum Einsatz, ausgestattet mit einem PAL-Autosampler (Agilent Technologies).
Die Detektion und Quantifizierung erfolgte im Multiple Reaction Monitoring (MRM)-Modus. Die spezifischen Ionenübergänge für die Zielanalyten sowie die verwendeten internen Standards sind in Tab. 4 dargestellt.
Detektierte Ionen am Gaschromatograph–Triple-Quadrupol-Massenspektrometer während der MRM-Phase
| Verbindung | Vorläuferion [m/z] | Produktion [m/z] |
|---|---|---|
| 4MSP-d10 | 228 | 46 |
| 4MSP-d10 | 110 | 46 |
| 4MSP | 218 | 43 |
| 4MSP | 100 | 43 |
| 3SHA-d5 | 265 | 73 |
| 3SHA-d5 | 205 | 59 |
| 3SHA | 260 | 55 |
| 3SHA | 200 | 59 |
| 3SH-d5 | 223 | 57 |
| 3SH-d5 | 178 | 55 |
| 3SH | 218 | 67 |
| 3SH | 173 | 61 |
Für die Festphasenextraktion (SPE) wurden 1-mL-Kartuschen mit 30 mg Lichrolut EN befüllt. Die Konditionierung der Säulen erfolgte schrittweise mit jeweils 1 mL tert-Butylmethylether, Methanol, deionisiertem Wasser, Tris-Puffer (0,1 M, pH 7,2) sowie 2 mL PHMB-Lösung (2 mM in 0,1 M Tris-Puffer, pH 7,2). Im Anschluss wurden 50 mL Weinproben, versetzt mit 50 μL eines internen Standard Mixes (Konzentration: 1 mg/L je Verbindung) und 2 mL gesättigter EDTA-Lösung (bei Raumtemperatur), langsam über die SPE-Säulen appliziert.
Die Säulen wurden anschließend mit 1 mL deionisiertem Wasser, 5 mL eines Methanol-Weinsäurepuffer-Gemisches (60:40, v/v; Puffer: 5 g/L, pH 3,0) sowie nochmals mit 1 mL deionisiertem Wasser gewaschen. Nach dem Trockensaugen wurde die Matrix mit 4 mL n-Pentan eluiert und erneut getrocknet.
Die Elution der gebundenen Thiole erfolgte in drei Schritten à 0,8 mL mittels einer Lösung von 7 mg/mL N-Acetylcystein in einem Lösungsgemisch aus Dimethylsulfoxid und tert-Butylmethyl ether (20:80, v/v). Das Eluat wurde in einem 20 mL Vial mit Schraubverschluss aufgefangen und mit 3,5 mL einer 1 %igen Natriumhydrogencarbonat-lösung versetzt, intensiv gemischt (Vortex) und zwischendurch belüftet. Nach Phasentrennung wurde die Etherphase abgehoben und dieser Schritt einmal wiederholt. Die kombinierten Ether-phasen wurden 2 Minuten bei 3000 U/min zentrifugiert und anschließend über Natriumsulfat getrocknet.
Für die Derivatisierung wurde das getrocknete Extrakt in ein frisches Schraubverschluss-Vial mit 0,6 mL eines 1:1-Gemisches aus Dimethylsulfoxid und Acetonitril überführt. Danach wurden 50 μL einer 10 %igen 1-Iodhexan-Lösung in tert-Butylmethylether sowie ca. 50 mg Cäsiumcarbonat zugegeben und durchmischt. Die Ansätze wurden 30 Minuten bei 60 °C im Heizblock inkubiert, anschließend kurz abgekühlt und ca. 10 mg Natriumborhydrid hinzugefügt. Nach erneutem Schütteln erfolgte eine zweite Inkubation bei 60 °C für weitere 30 Minuten.
Die Reaktionsansätze wurden nach dem Abkühlen mit 0,6 mL Isohexan versetzt, mit 3 mL deionisiertem Wasser gewaschen und 2 Minuten bei 3000 U/min zentrifugiert. Die obere organische Phase wurde abgenommen, dieser Waschvorgang wiederholt und die vereinigte organische Phase mit Natriumsulfat getrocknet. Anschließend wurde das Lösungsmittel in graduierten Glasröhrchen unter leichtem Stickstoffstrom bei 35 °C im Wasserbad bis auf ein Volumen von ca. 0,3 mL eingedampft. Die aufkonzentrierten Proben wurden in Mikrovials überführt und für die GC-MS/MS-Analyse bereitgestellt.
Die Kalibration erfolgte in Modellwein („Kunstwein“: 4 g/L Weinsäure, 12 % Ethanol, pH 3,2), in den definierte Konzentrationen der Zielanalyten eingearbeitet wurden. Der Kunstwein wurde aus deionisiertem Wasser hergestellt. Die Kalibrier standards wurden identisch zu den Proben extrahiert und derivatisiert. Zur Bestimmung der Wiederfindungsraten wurden fünf unterschiedliche Weine mit 10 ng/L und 50 ng/L 4MSP bzw. mit 50 ng/L und 500 ng/L 3SHA und 3SH versetzt. Die Reproduzierbarkeit der Methode wurde anhand von vier verschiedenen Weinen in fünffacher Wiederholung überprüft.
Die Analyse der Zielverbindungen erfolgte mittels Gaschromatographie gekoppelt mit Tandem-Massenspektrometrie (GC-MS/MS) im Large Volume Injection-Modus (LVI). Das Injektionsvolumen betrug 60,0 μL (Solvent-Split-Modus). Der Purge Flow zum Splitventil wurde nach 1 Minute auf 50 mL/min eingestellt.
Als Trennsäule kam eine ZB-FFAP-Säule mit einer Länge von 30 m, einem Innendurchmesser von 0,25 mm und einer Filmdicke von 0,25 μm zum Einsatz (Phenomenex Inc., California, USA). Helium diente als Trägergas mit konstantem Fluss bei 1,2 mL/min.
Das Temperaturprogramm des PTV-Injektors war wie folgt eingestellt: initial 45 °C für 12 Sekunden, anschließend Aufheizung auf 250 °C mit einer Rate von 500 °C/min; keine Haltezeit am Endpunkt.
Das Ofentemperaturprogramm gliederte sich wie folgt:
Initiale Temperatur: 45 °C (5 min Haltezeit)
Aufheizung auf 110 °C mit 5 °C/min (keine Haltezeit)
Weiterer Temperaturanstieg auf 230 °C mit 9 °C/min (keine Haltezeit)
Endtemperatur: 250 °C mit 15 °C/min und einer Haltezeit von 7 Minuten
Die Quantifizierung erfolgte anhand der Peakflächen unter Verwendung eines internen Standards: 4MSP-d10, 3SH-d5 und 3SHA-d5 wurden als isotopenmarkierter Mixstandard eingesetzt.
Die entwickelte Analysemethode erwies sich als hochleistungsfähig und zuverlässig für die Quantifizierung der drei Zielverbindungen 4MSP, 3SH und 3SHA im Konzentrationsbereich von 10 bis 500 ng/L. Die Kalibrierkurven zeigten durchgehend eine exzellente Linearität (R2 > 0,99), wie in den Abb. 1 bis 3 dargestellt. Die Wiederfindungsraten von 87,8 % bis 126,7 % (Tab. 5) liegen im akzeptablen Bereich für Methoden mit komplexer mehrstufiger Probenvorbereitung. Abweichungen von über 120 % können durch Matrixeffekte oder geringe Variationen im Derivatisierungsschritt erklärt werden. Die geringen relativen Standardabweichungen (RSD: 0,88 %–6,64 %, Tab. 6) belegen die hohe Präzision und analytische Genauigkeit der Methode. Diese Werte decken sich mit der Literatur (vgl. Coetzee et al., 2018).
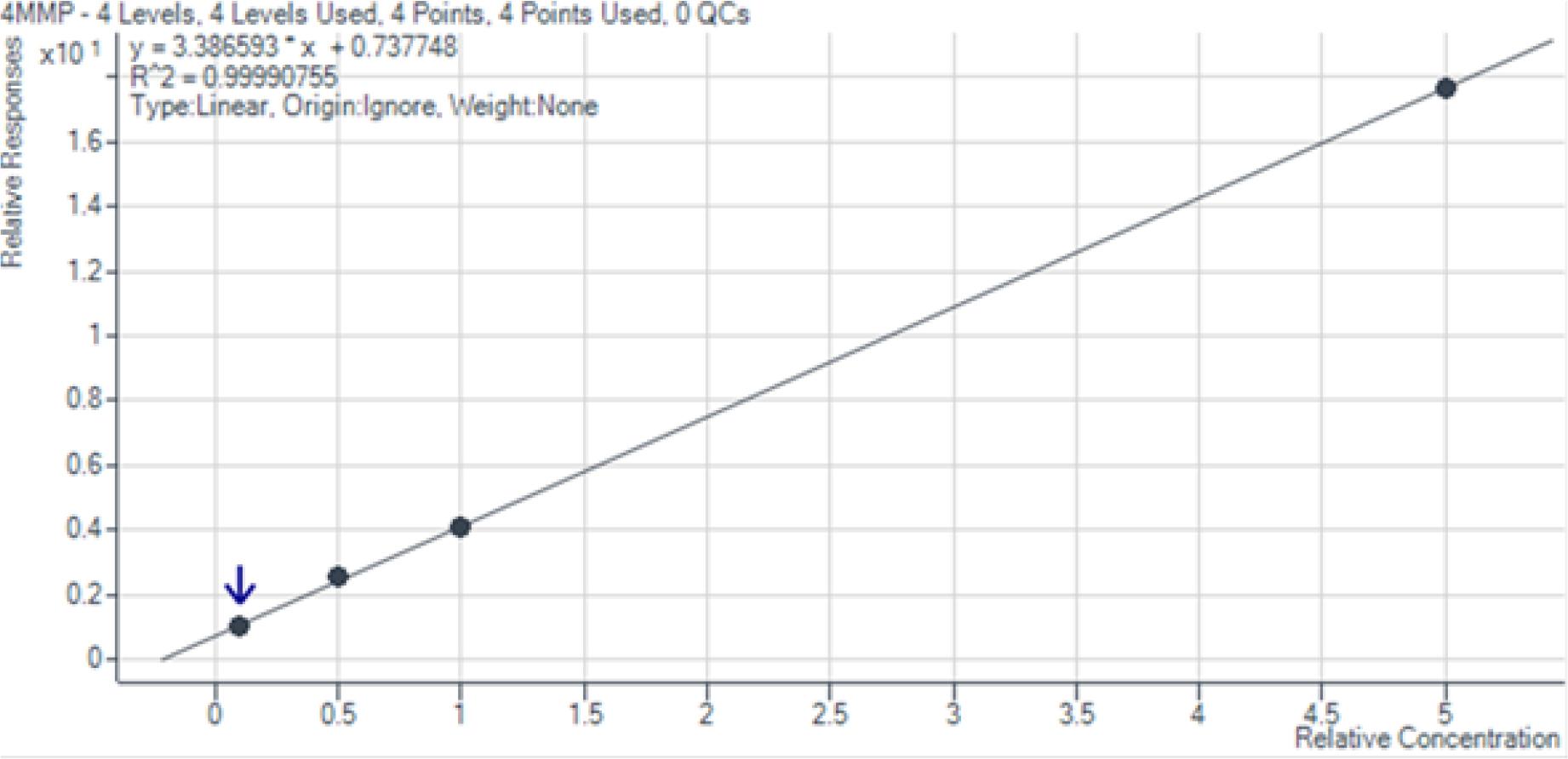
Kalibrationskurve 4-Methyl-4-sulfanylpentan-2-on im Bereich von 10 bis 500 ng/l
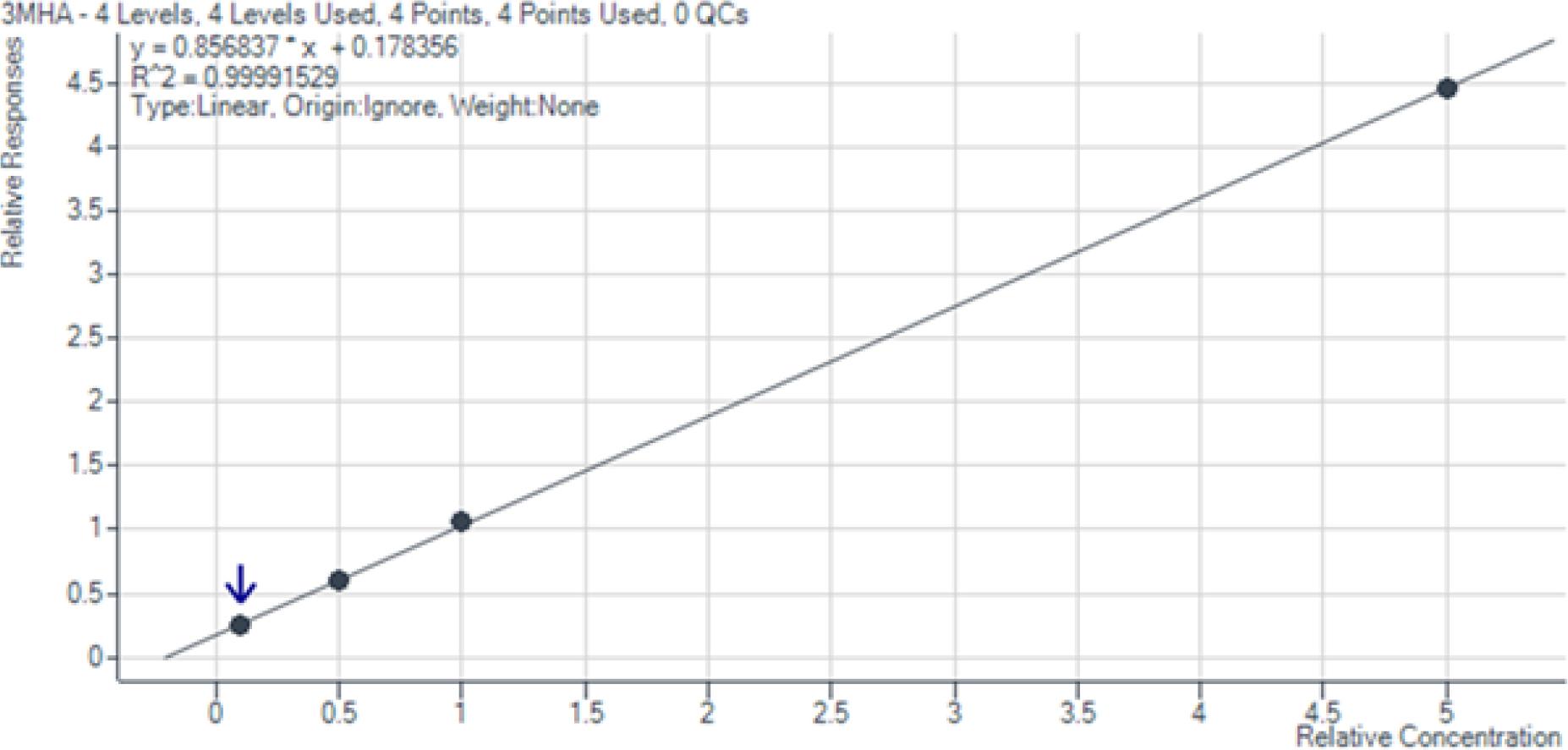
Kalibrationskurve 3-Sulfanylhexylacetat im Bereich von 10 bis 500 ng/l
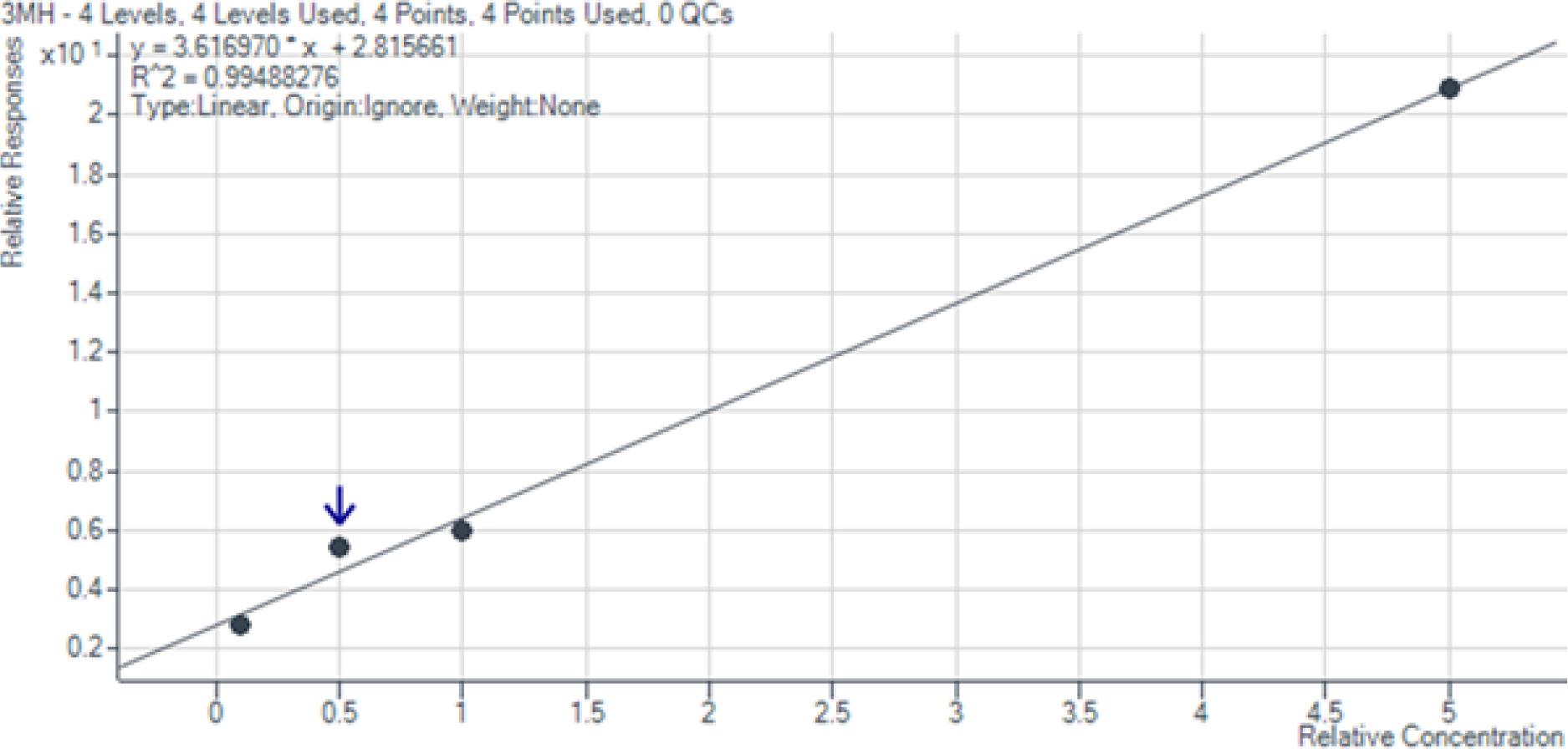
Kalibrationskurve 3-Sulfanylhexan-1-ol im Bereich von 10 bis 500 ng/l
Wiederfindungsrate in unterschiedlichen Weinen
| 4MSP | 0-Probe [ng/l] | Zusatz von 10 ng/l [ng/l] | Wiederfindung % | Zusatz von 50 ng/l [ng/l] | Wiederfindung % |
|---|---|---|---|---|---|
| Probe 1 | n.d. | 12,1 | 121,0 | 48,2 | 96,5 |
| Probe 2 | n.d. | 9,8 | 98,1 | 50,0 | 100,1 |
| Probe 3 | n.d. | 12,7 | 126,7 | 47,1 | 94,2 |
| Probe 4 | n.d. | 9,9 | 99,0 | 48,5 | 96,9 |
| Probe 5 | n.d. | 8,8 | 87,8 | 46,8 | 93,5 |
| 3SHA | 0-Probe [ng/l] | Zusatz von 50 ng/l [ng/l] | Wiederfindung % | Zusatz von 500 ng/l [ng/l] | Wiederfindung % |
| Probe 1 | 3,4 | 53,3 | 99,9 | 517,6 | 102,8 |
| Probe 2 | n.d. | 52,0 | 104,1 | 503,2 | 100,6 |
| Probe 3 | n.d. | 53,4 | 106,8 | 512,6 | 102,5 |
| Probe 4 | 3,4 | 55,7 | 104,6 | 486,8 | 96,7 |
| Probe 5 | 2,6 | 53,4 | 101,6 | 489,1 | 97,3 |
| 3SH | 0-Probe [ng/l] | Zusatz von 50 ng/l [ng/l] | Wiederfindung % | Zusatz von 500 ng/l [ng/l] | Wiederfindung % |
| Probe 1 | n.d. | 50,3 | 100,6 | 504,3 | 100,9 |
| Probe 2 | n.d. | 54,8 | 109,6 | 482,9 | 96,6 |
| Probe 3 | n.d. | 55,1 | 110,2 | 518,0 | 103,6 |
| Probe 4 | n.d. | 47,7 | 95,4 | 503,3 | 100,7 |
| Probe 5 | n.d. | 56,3 | 112,6 | 506,6 | 101,3 |
Reproduzierbarkeit in unterschiedlichen Weinen
| Wf1% | Wf2% | Wf3% | Wf4% | Wf5% | Rel.StdAbw % | ||
|---|---|---|---|---|---|---|---|
| WW 1 | 4MSP Results [ng/l] | 14 | 15 | 13 | 13 | 14 | 6,64 |
| 3SHA Results [ng/l] | 41 | 44 | 43 | 40 | 40 | 4.53 | |
| 3SH Results [ng/l] | 278 | 271 | 285 | 281 | 288 | 2,34 | |
| WW 2 | 4MSP Results [ng/l] | 22 | 19 | 20 | 20 | 20 | 6,21 |
| 3SHA Results [ng/l] | 134 | 138 | 129 | 136 | 128 | 3,24 | |
| 3SH Results [ng/l] | 117 | 126 | 120 | 123 | 119 | 3,03 | |
| RW 1 | 4MSP Results [ng/l] | 14 | 14 | 13 | 14 | 12 | 5,09 |
| 3SHA Results [ng/l] | 121 | 123 | 120 | 118 | 121 | 1,42 | |
| 3SH Results [ng/l] | 167 | 172 | 158 | 165 | 164 | 3,01 | |
| RW 2 | 4MSP Results [ng/l] | 50 | 52 | 53 | 52 | 55 | 3,47 |
| 3SHA Results [ng/l] | 208 | 206 | 206 | 205 | 203 | 0,88 | |
| 3SH Results [ng/l] | 205 | 207 | 198 | 204 | 201 | 1,74 |
Bereits während der Methodenetablierung wurde ersichtlich, dass existierende Ansätze aus der Fachliteratur – insbesondere PFB-bzw. Propionat-Derivatisierungen nach Mateo-Vivaracho et al. (2007), Rodríguez-Bencomo et al. (2009) und Musumeci et al. (2015) – nicht ohne Weiteres auf die in dieser Untersuchung betrachteten Matrizes, Konzentrationsbereiche und Geräteeinstellungen übertragbar waren. Somit wurden mehrere zentale Prozessschritte gezielt weiterentwickelt. Dazu gehörte unter anderem die Auswahl und Vorbehandlung der SPE-Kartuschen sowie die Optimierung der Elutionsbedingungen, bei welcher sich eine Lösung von N-Acetylcystein in DMSO/MTBE als wirkungsvoll erwies. Außerdem wurden die Bedingungen der Iod-Derivatisierung systematisch untersucht und angepasst, da die in der Literatur beschriebenen Parameter (u. a. Musumeci et al., 2015) in Weinmatrix nicht die erforderliche Reproduzierbarkeit zeigten. Darüber hinaus wurde ein reduktiver Nachbehandlungsschritt mit Natriumborhydrid implementiert, welcher die Signalstabilität und Peakform im MRM-Modus signifikant verbesserte. Darüber hinaus mussten die LVI- und Solvent-Vent-Parameter des Injektors sowie der MRM-Übergang im Triple-Quadrupol-MS optimiert werden, um ein maximales Signal-zu-Rausch-Verhältnis zu erzielen. Diese Prozessschritte ermöglichen den wesentlichen methodischen Fortschritt der vorliegenden Arbeit und bilden die Grundlage für die analytischen Kennzahlen.
Die Nachweisgrenzen (Limit of Detection, LOD) sowie die Quantifizierungsgrenzen (Limit of Quantification, LOQ) wurden gemäß dem Signal-Rausch-Verhältnis (S/N) bestimmt, wobei ein Verhältnis von 3:1 für die LOD und 10:1 für die LOQ zugrunde gelegt wurde. Die berechneten LODs lagen bei 6,8 ng/L für 4MSP, 5,9 ng/L für 3SHA und 9,3 ng/L für 3SH; die entsprechenden LOQs bei 22,7 ng/L (4MSP), 19,6 ng/L (3SHA) und 31,0 ng/L (3SH). Diese Ergebnisse unterstreichen die hohe Sensitivität der Methode für die Detektion flüchtiger schwefelhaltiger Aromakomponenten im niedrigen ng/L-Bereich. Musumeci et al. (2015) beschrieben die Nachweisgrenzen für 4-MSP von 0,9 ng/L, für 3-SH 1 ng/L bzw. 3-SHA 17 ng/L unter Verwendung einer HS-SPME-basierten Derivatisierung mit Propionat-Reagenzien. Mateo-Vivaracho et al. (2007) erreichten mit einer PFB-Derivatisierung und negativer chemischer Ionisation noch niedrigere Nachweisgrenzen von 0,1 ng/L für 4-MSP, 0,6 ng/L für 3-SHA und 7 ng/L für 3-SH, was jedoch mit dem Einsatz halogenierter Reagenzien und längeren Aufarbeitungszeiten verbunden war.
Zur besseren Einordnung wurde eine Tabelle mit detaillierten Ergebnissen zu den bisherigen Methoden der Analyse von varietalen Thiolen gegenübergestellt (siehe Tab. 7). Die in der Fachliteratur beschriebenen Methoden basieren in der Regel auf SPME-, SBSE-oder halogenierten Derivatisierungsreagenzien wie PFBBr. Obwohl diese Reagenzien teilweise eine geringere LOD ermöglichen, sind sie mit einem höheren Materialaufwand, empfindlichen Spezialkomponenten oder dem Umgang mit toxikologisch kritisch eingestuften Reagenzien verbunden. Die in dieser Arbeit eingesetzte Kombination aus Festphasenextraktion und Iod-Derivatisierung nutzt ausschließlich konventionelle Lösungsmittel und schließt somit halogenierte oder reaktive Reagenzien aus. Dies führt zu einer verbesserten Arbeitssicherheit und ökologischen Bewertung. Gleichzeitig liegen die erzielten Nachweisgrenzen im Bereich anderer etablierter Verfahren, wodurch die Methode eine praxistaugliche und ressourcenschonende Alternative darstellt.
Vergleich ausgewählter analytischer Methoden zur Bestimmung varietaler Thiole
| Studie / Plattform | Probenvorbereitung / Derivatisierung | Detektion | LOD (ng/L) |
|---|---|---|---|
| Diese Arbeit | SPE-Extraktion; Derivatisierung mit 1-Iodhexan & Cs2CO3; LVI-PTV | GC-MS/MS (MRM) | 6,8 / 9,3 / 5,9 |
| Mateo-Vivaracho et al. (2007) | LLE + PFBBr-Derivatisierung | GC-NICI-MS | 0,1 / 7 / 0,6 |
| Rodríguez-Bencomo et al. (2009) | HS-SPME + PFBBr-Derivatisierung | GC-MS | < Geruchsschwelle |
| Musumeci et al. (2015) | Extractive Alkylation + HS-SPME | GC-EI-MS | 0,9 / 1 / 17 |
| Ochiai & Kishimoto (2015) | der-SBSE (Twister) | TD-GC-MS/MS | 0,20 / 27 / 0,19 |
Das hier angewandte Verfahren zeichnet sich im Vergleich zu den in der Literatur beschriebenen Methoden, wo eine direkte Derivatisierung oder adsorptive Extraktionstechniken erfolgen, durch eine deutlich verbesserte Matrixreinigung mittels Festphasenextraktion und anschließender Iod-Derivatisierung aus, welches eine weitgehende Eliminierung von Matrixinterferenzen und eine sehr gute Reproduzierbarkeit erlaubt.
Zusätzlich weist die entwickelte Methode mehrere praktische und ökologische Vorteile gegenüber bestehenden Verfahren auf. Der Arbeitsablauf ist auf eine routinetaugliche Anwendung im weinanalytischen Labor ausgelegt und kommt ohne spezielle und besonders teure Materialien wie SPME-Fasern aus. Durch den Einsatz isotopenmarkierter interner Standards lassen sich Matrixeffekte kompensieren, was zu hoher Reproduzierbarkeit der Messungen beiträgt. Auch in zeitlicher und apparativer Hinsicht erweist sich das Verfahren als effizient: Der Extraktions- und Derivatisierungsablauf nimmt weniger als zwei Stunden in Anspruch und kann weitgehend automatisiert werden. Da ausschließlich gängige Lösungsmittel wie DMSO und Acetonitril verwendet werden und vollständig auf halogenierte Reagenzien verzichtet wird, entspricht die Methode zudem in hohem Maß den Prinzipien der „Green Chemistry“. Durch den Wegfall von NICI-bzw. PFB-basierten Ansätzen entsteht kein Risiko durch toxische oder schwer entsorgbare Chemikalien, welches das ökologische Profil der Methode wesentlich verbessert. Die im niedrigen ng/L-Bereich liegenden LOD- und LOQ-Werte zeigen, dass die erzielte Empfindlichkeit für die Routinebestimmung in Weinproben mehr als ausreichend ist, während der analytische Aufwand gleichzeitig moderat bleibt. In Summe steht somit eine praxistaugliche und nachhaltige Alternative zu anderen Verfahren zur Verfügung.
Obwohl die Geruchsschwelle von 4MSP bei etwa 0,8 ng/L liegt (Coetzee et al., 2018), können auch höhere Konzentrationen im zweistelligen ng/L-Bereich sensorisch und analytisch relevant sein. Im Vergleich zu früheren Verfahren mit ähnlicher Derivatisierungsstrategie (z. B. Herbst-Johnstone et al., 2013) bietet die hier eingesetzte GC-MS/MS-Methode eine gesteigerte Empfindlichkeit und verbesserte Nachweisleistung, insbesondere hinsichtlich der Robustheit gegenüber Matrixeffekten.
Die Analyse der Weinproben ergab eine ausgeprägte rebsortenspezifische Verteilung der Thiolverbindungen. In Weinen der Rebsorte Grüner Veltliner wurden mit wenigen Ausnahmen nur geringe Mengen an 4MSP detektiert. 3SHA konnte hingegen in sämtlichen Proben nachgewiesen werden (19,2–310,9 ng/L), während 3SH durchgängig in vergleichsweise hohen Konzentrationen vorlag (85,0–385,3 ng/L). Diese Befunde weisen auf ein grundsätzliches Potenzial zur Bildung thiolbasierter Aromen auch außerhalb der klassischen Sauvignon-Blanc-Typizität hin.
In Sauvignon Blanc-Weinen wurden erwartungsgemäß besonders hohe Konzentrationen von 4MSP gemessen (bis zu 24,4 ng/L), was das für diese Rebsorte charakteristische, vegetabiltropische Aromaprofil bestätigt (vgl. Coetzee et al., 2012). Auch 3SHA (115,7–183,8 ng/L) und 3SH (89,3–194,7 ng/L) waren in sämtlichen Proben in signifikanten Mengen vorhanden.
Der Gemischte Satz, eine aus mehreren weißen Rebsorten zusammengestellte Kategorie, zeigte eine große intraindividuelle Variabilität. 4MSP wurde nur in einzelnen Proben über der Nachweisgrenze quantifiziert (bis 16,6 ng/L). 3SHA konnte – mit einer Ausnahme – in allen Proben detektiert werden (24,7–181,3 ng/L), während insbesondere vier Weine durch außergewöhnlich hohe 3SH-Gehalte (>300 ng/L) auffielen.
In Blaufränkisch-Weinen konnte 3SHA in keiner der untersuchten Proben nachgewiesen werden, während 4MSP in Spuren bis zu 32,0 ng/L vorkam. Bemerkenswert war die durchgängig hohe Präsenz von 3SH (220,8–379,1 ng/L), was auf die potenzielle Bedeutung dieser Verbindung für das Aromaprofil auch in Rotweinen hinweist.
Die Rebsorte Zweigelt zeigte konsistent nachweisbare Konzentrationen von 4MSP (2,5–27,9 ng/L) und 3SHA (117,3–185,8 ng/L). Die 3SH-Gehalte schwankten dagegen deutlich (86,8–258,1 ng/L), was auf vinifikatorische Einflüsse wie Reifegrad, Hefestamm oder Gärführung schließen lässt.
In den aus Blauer Wildbacher hergestellten Schilcher-Roséweinen wurden teilweise sehr hohe 3SH-Konzentrationen (>300 ng/L) gemessen. Die 4MSP-Werte lagen zwischen 0,0 und 10,9 ng/L, während 3SHA stark schwankte (0,0–217,4 ng/L).
Insgesamt belegen die Ergebnisse die ausgeprägte Rebsortenabhängigkeit flüchtiger Thiolverbindungen im Wein und unterstreichen zugleich die Bedeutung vinifikatorischer Strategien für die gezielte Steuerung des Aromapotenzials. Die hohe Sensitivität und Reproduzierbarkeit der Methode macht sie zu einem geeigneten Instrument für den Einsatz in der weinaromatischen Forschung und der qualitätsorientierten Kellerwirtschaft.
Zur Veranschaulichung der chromatographischen Trennung wurden exemplarisch zwei Chromatogramme ausgewählter Weine beigefügt (Abb. 4 und 5).
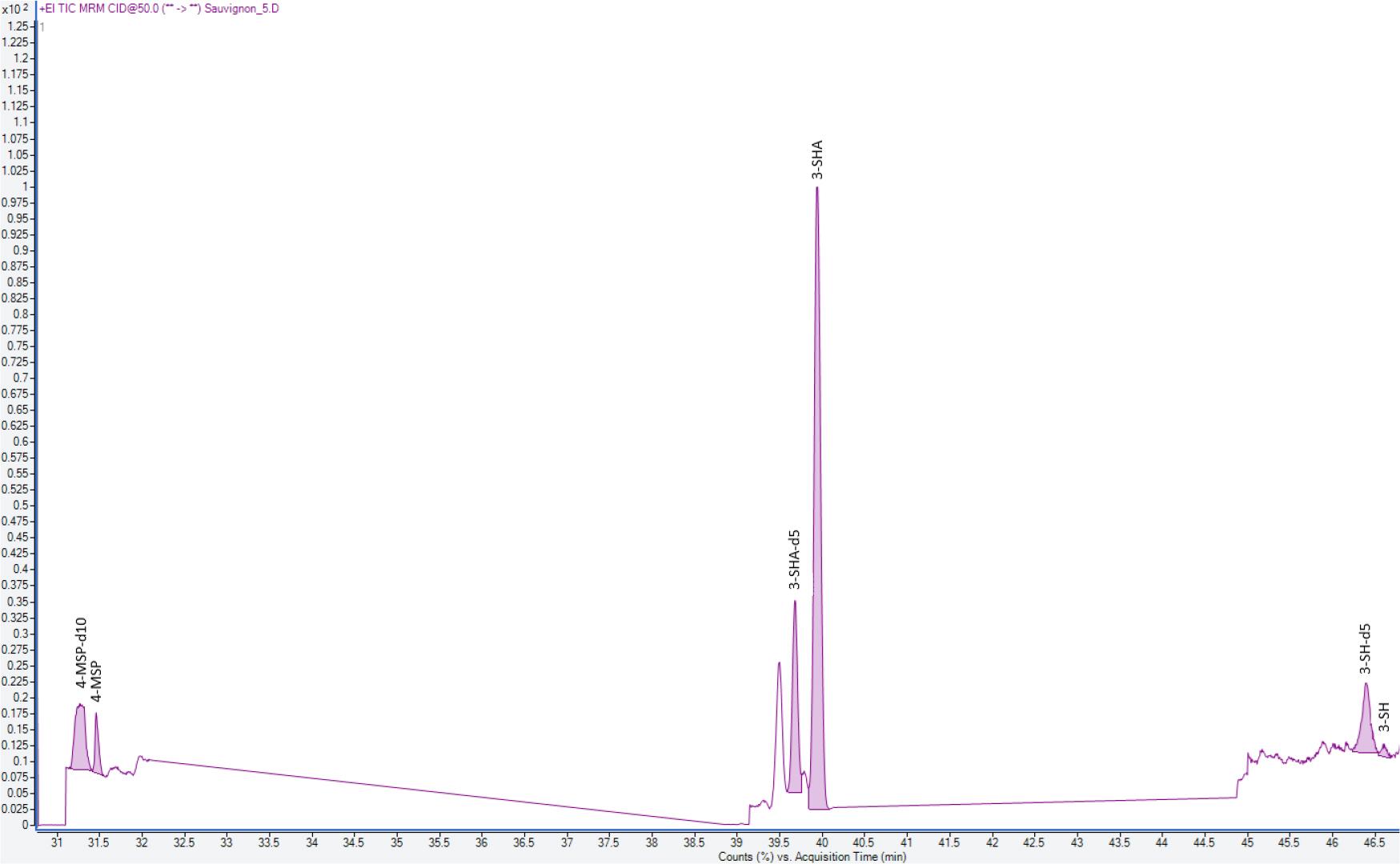
Chromatogramm aus einem Sauvignon Blanc-Wein
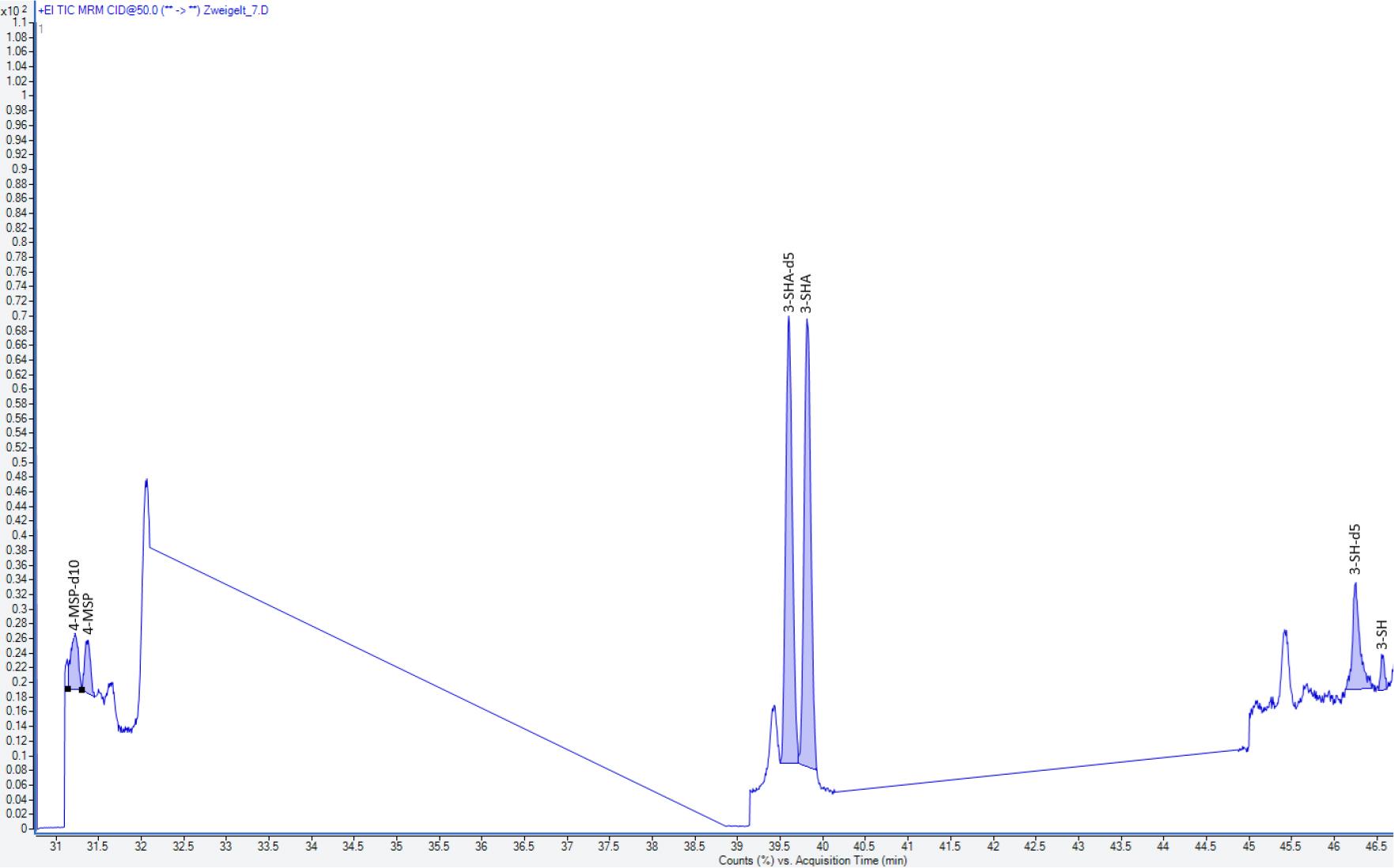
Chromatogramm aus einem Zweigelt-Wein
Für die wertvollen Hinweise und die sorgfältige Durchsicht im Rahmen der finalen Manuskriptanpassung danke ich Herrn Nikolaus Schlögl, M.Sc. M.Ed., sehr herzlich.