
Optical genome mapping (OGM) is a novel technology enabling the detection of structural genomic variants (SV) at a resolution and in size range previously not readily available by other methods, opening new fields of research1,2. In human diagnostics, OGM has so far been applied to cancer genetics / haematology3–6, constitutional molecular genetics1,2, quality control assurance in genome modification (such as detection of off-target effects in genetically modified cell lines)7, and in routine clinical genomic diagnostics of facioscapulohumeral dystrophy (FSHD)8–11.
OGM has been in use at the Clinical Institute of Genomic Medicine (CIGM), University Medical Centre Ljubljana (UMCL), Slovenia since 2021 for research and diagnostic purposes. Our research focus (ARIS Programme P3-0326) involves discovering mechanisms of unexplained recurrent spontaneous pregnancy loss, male infertility, and integration and co-interpretation of whole genome sequencing (WGS) and OGM data (ARIS Programme J3-4517). The planned integration of OGM data with WGS will hopefully further increase the yield of diagnostics in such cases. In routine genetic diagnostics at CIGM, OGM is currently used for diagnostic testing of FSHD11, characterization and resolution of variants identified by other technologies, and undiagnosed rare disease patients. In this way, we have so far successfully used OGM to characterize variants identified by other technologies, such as microarray, NGS, and karyotyping, to resolve the clinical significance of various SVs12.
Herein, we present a family case report of rare disease patients tested using OGM that was performed in collaboration with the Center for Medical Genetics and Immunology (CMGI), Clinical Center of Montenegro (CCM) (BI-ME/21-22-016). In addition to the challenges faced in interpretation of SVs based on strictly applied ACMG criteria and ClinGen guidelines, our work serves to highlight the complexity of the diagnostic journey in rare disease cases.
Two undiagnosed male siblings with an overlapping clinical presentation of thrombocytopenia, sacro-coccy-geal teratoma, hydronephrosis/reflux vesicoureteral and obesity, who were referred to the CGMI, CCM, Montenegro, were enrolled in this family case-report.
Clinical data was collected during the patients’ inperson appointments and evaluation by clinical geneticist at the CGMI, CCM, and all specialist examinations, were performed as part of standard routine clinical care. Before genetic testing, pre-test genetic counseling was provided by a clinical geneticist, followed by obtaining written parental consent at the CGMI, CCM. All procedures in the study were conducted according to the routine standard of care and in accordance with the principles of the Declaration of Helsinki. Karyotyping was performed at the CGMI, CCM, while microarray analyses, exome sequencing and optical genome mapping were performed at the CIGM, UMCL, Slovenia.
Chromosome analysis was performed for both probands, by using G-bended karyotyping (bend resolution 400–470, according to ISCN), after 72 hours of peripheral blood cultivation.
Microarray analysis was initially performed on the probands and their parents by using oligonucleotide array Agilent Technologies 4×180K (AMADID:035689), according to the manufacturer’s instructions. Agilent Cy-toGenomics 5.1.2.1 software was used to visualize and report the CNVs, as previously described 12.
Exome sequencing of proband 1 and proband 2 with parents in trio setup was performed as previously described 13,14, and included the analysis of a total of >2000 genes associated with the clinical phenotype of the probands. The full list of genes for each of the included gene panels is available in the Supplement.
Optical genome mapping was performed as previously described12. Briefly, high-weight molecular DNA was extracted from 1.5 million lymphocytes from whole blood (EDTA collected) using the SP Blood & Cell Culture DNA Isolation Kit or the SP-G2 Blood & Cell Culture DNA Isolation Kit following manufacturer instructions (Bionano Genomics Inc., San Diego USA). The following day, DNA molecules were labeled with the DLE-1 enzyme using the Direct Label and Stain (DLS) Kit or Direct Label and Stain-G2 (GLS-G2) kit (Bionano Genomics Inc.). Labeled DNA was loaded on the three-flowcell Saphyr Chip® G2.2 or G2.3 (Bionano Genomics Inc.) and ran on the Saphyr instrument (Bionano Genomics Inc.) to reach a minimum yield of 500 Gbp (DLE-1 label, [GRCh38] reference genome). The de novo assembly and Variant Annotation Pipeline were executed on Bionano Solve3.7_20221013_25 while reporting and direct visualization of SVs was done on Bionano Access 1.7.2.
We reported only those genomic variants that have statistical support based on the adequate genomic coverage and chosen analysis type for the detection of CNV, duplications, deletions, and other SVs such as insertions, inversion, intra- and inter-chromosomal translocations, as determined by internal Access QC parameters. The method does not enable the analysis of regions that do not contain DLE-1 labeling sites (centromeres, telomeres, and other heterochromatin regions). According to the ACMG and ClinGen guidelines15, CNV variants are classified into one of five classes of pathogenicity based on the sum of points in each category of assessment, and were classified by comparison with their overlap with SV and CNV variants contained in the DGV (Database of Genomic Variants - http://dgv.tcag.ca/gb2/gbrowse/dgv2_hg19/)16, gnomAD (genome Aggregation Database - https://gno-mad.broadinstitute.org/), OGM (Bionano Genomics Inc. internal Access® database), ClinGen (Clinical Genome Resource Consortium) (https://dosage.clinicalgenome.org/), DECIPHER (https://www.deciphergenomics.org/), and/or ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) public databases and the CIGM genomic variant database. OGM results are given according to the genome mapping nomenclature as specified in the ISCN guidelines17.
Figure 1 was prepared from original visualizations generated by Bionano Access 1.7.2 software (Bionano), segregation, and optical genome mapping, respectively. The final composite Figure 1 was technically prepared in terms of size, layout, format, and type of file with no modification to the original data, from the original visualizations, by using GIMP 2.1018. The pedigree was constructed and drawn using (Progeny Clinical Version N/Progeny Lab Version N) (Progeny Genetics LLC, Aliso Viejo, CA, www.progenygenetics.com).
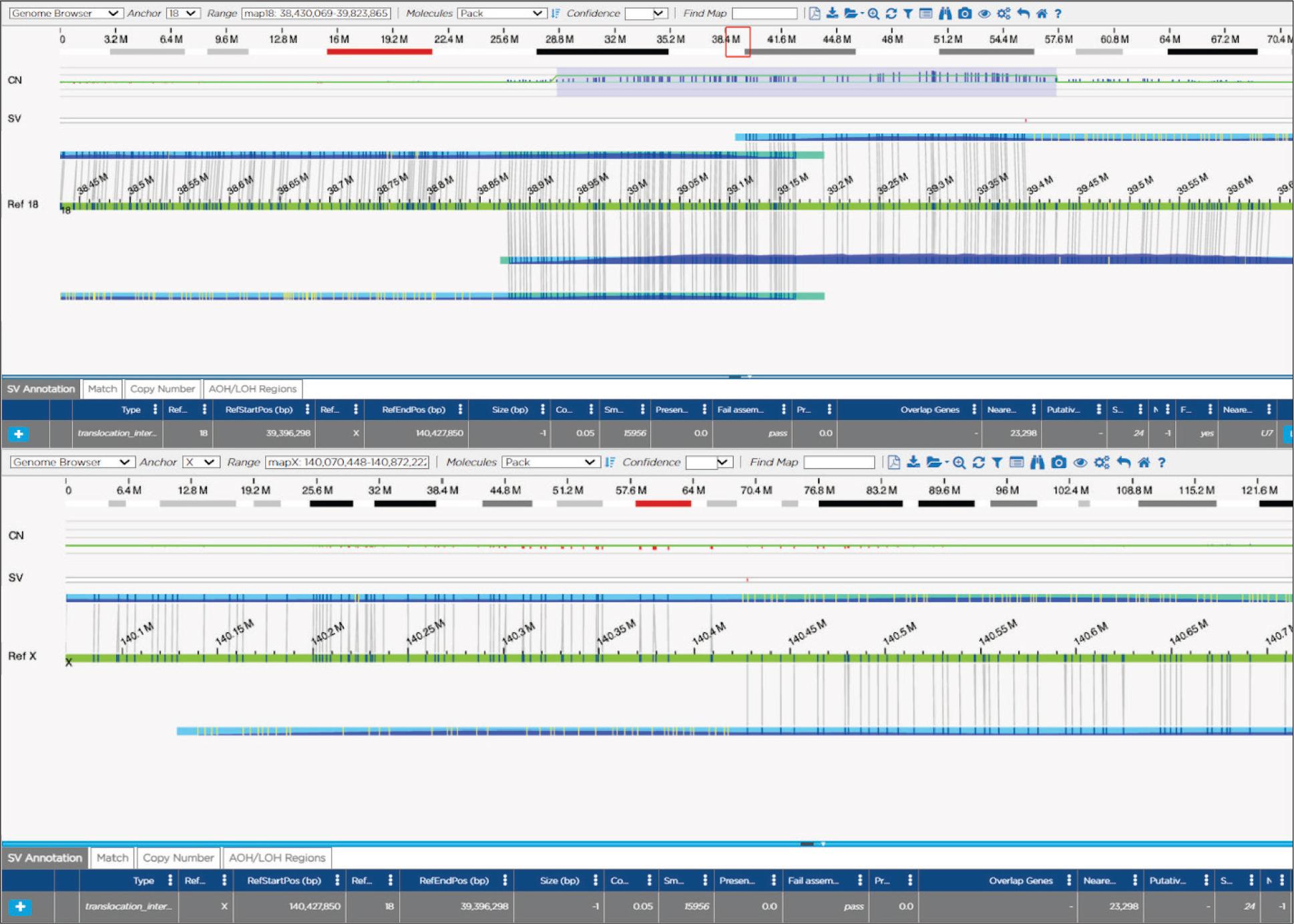
Optical genome mapping results showing molecules involved in the translocation mapping to chromosome 18 and chromosome X.
Characteristics of two male siblings (proband 1 and proband 2) with an overlapping clinical presentation of thrombocytopenia, sacrococcygeal teratoma, hydrone-phrosis/reflux vesicoureteral and obesity, are shown in Table 1.
Clinical characteristics of the probands
| Clinical characteristics | Proband 1 | Proband 2 | Onset |
|---|---|---|---|
| Age at first visit | 11 yr. & 11 mo. | 3yr. & 6 mo. | |
| Early development | Hyperactivity | Normal | |
| Teratoma regio sacro-coccigealis | + | + | Neonatal |
| Hydronephrosis / *RVU | + (right side) | + (left side) | Neonatal |
| Thrombocytopenia | + | + | Neonatal |
| Purpura, petechiae, bruises | + | + | Infancy |
| Premaxillary prominence | + | + | Toddler |
| Juvenile palmoplantar dermatosis | + | + | Toddler |
| Obesitas | + | + | Toddler |
| Hypo-imunoglobulinaemia | + | 10 yr. | |
| Ameloblastoma mandibulae | + | 10 yr. | |
| Intellectual disability, mild | + | 6 – 7 yr. | |
| Epilepsia | + | 14 yr. | |
| Cerebral dysmyelination (MRI) | + | 17 yr. | |
| Frontoparietal polymicrogyria (MRI) | + | 17 yr. |
*Reflux vesico-ureteral
Both parents and three sisters of the probands were healthy and without any of the clinical signs and symptoms shown in the probands, except for a few asymptomatic episodes of borderline platelet values in the mother.
Normal male karyotypes were detected for both male siblings (Proband 1: 46, XY; Proband 2: 46, XY), with no clonal abnormalities (30 metaphases), at the stated band level of resolution. Karyotypes of the parents were also normal (Mother: 46, XX; Father: 46, XY).
Microarray analyses of the proband 1 showed an interstitial single copy gain of 18q12.2 region, approximately 459,7 kb in size in a male profile: arr[GRCh38] 18q12.2(38880911_39340584)×3 (arr[GRCh37] 18q12.2(36460875_36920548)×3). The identified duplication did not overlap with any known disease-causing genes and was not present in the databases containing variants from healthy individuals (DGV), nor in the medical literature or databases ClinGen, ClinVar, or DECIPHER. Due to its size and rarity, the copy number gain was interpreted as a variant of unknown significance, and segregation analysis using arrays was recommended. Segregation testing using microarrays in the mother and father of the proband showed the presence of the same 18q12.2 copy number gain in the mother of the proband. As the molecular karyotyping showed the presence of the same interstitial duplication of the 18q12.2 region in the proband and the mother; arr[GRCh38] 18q12.2(38880911_39340584)×3m at, and the duplication did not affect clinically important genes, it was interpreted as a likely benign genomic variant, and so clinical testing was continued to determine the cause of the clinical presentation in the proband and his brother.
Exome sequencing was initially performed for proband 1, as previously described13,14. The original gene panel included >100 genes associated with thrombocytopenia and hereditary thrombocytopenia including Wiskott-Aldrich syndrome. The analysis did not identify any variants that could explain the phenotype and therefore a reinterpretation of the exome sequencing data of proband 1 was performed with an expansion to genes associated with the additional phenotypes observed (Table 1). Despite adding over 1500 genes to the analysis, no causative variants were identified. Finally, exome sequencing in trio setup was performed for proband 2 and both parents with updated gene panels. Despite including >2000 genes associated with the clinical phenotypes, no conclusively causative SNV variants or small indels could be identified. While the duplication observed on the microarray analysis was apparent from the coverage, no breakpoints could be identified by exome sequencing. The full list of genes included in the exome sequencing analysis is available in the Supplement.
Optical genome mapping showed a translocation between chromosomes X and 18, accompanied by a duplication of the 18q12.2 region, of maternal origin in both probands; ogm[GRCh38] t(X;18)(q27.1;q12.2)(140408784~140427850;38878133~39396298)mat,dup(18) (18q12.2)(38927193_39426970)mat. The translocation breakpoints and the associated duplication of the 18q12.2 region did not overlap any clinically significant genes and are unlikely to be visible using classic karyotyping methods. The accompanying 18q12.2 duplication was approximately 499,7 kb in size, and was consistent with the previously observed duplication in the proband 1, 2 and their mother using microarray analysis: arr[GRCh38] 18q12.2(38880911_39340584)×3mat (Figure 1).
The translocation and accompanying duplication of maternal origin do not directly affect genes known to cause disease in humans. However, several genetic mechanisms are known to influence the expression of nearby genes by influencing regulatory regions or by topological means, some promoting and some inhibiting expression 19–22. Therefore, as described previously, we used the UCSC Genome Browser Viewer to visualize our region of interest in the context of neighboring genomic regions 12,23, however no obvious regulatory regions explaining the phenotype could be identified as being affected by the detected translocation and accompanying duplication. However, literature search revealed that the region of chromosome 18 involved in the rearrangement has previously been described in the context of germinal translocation t(11;18) (q22.1;q12.2), (ClinVar ID: 599287), where the translocation carriers also had age-dependent hypertension linked to 11q22.1, as well as obesity 24. Additionally, somatic translocations between chromosome X and chromosome 18 involving different breakpoints have been previously described in synovial sarcomas (t(X;18)(p11.2;q11.2)), including in a rare case with submandibular presentation 25,26, however, the exact breakpoints of the critical region of 18q11.2 do not correspond to those identified in our patients. Therefore, because of the lack of direct evidence of pathogenicity, but because of the clinical match of the probands, the involvement of the chromosome X in males and a female with a very slight phenotype of transient thrombocytopenia, and the size of regions possibly affected indirectly, the translocation was classified as a variant of unknown clinical significance. When we are unable to provide final conclusions, extensive segregation may prove beneficial to clarifying the classification of the variant, as recently described in case of a PLP1 duplication by our group12. In case of variants involving chromosome X in males, testing additional male family members may provide additional information helpful to clinical clarification, which is why we expanded the segregation to include healthy brothers of the carrier mother. The results of the segregation analysis are shown in Figure 2. None of the four maternal uncles were carriers of this rare familial translocation, that remains a variant of unknown significance.
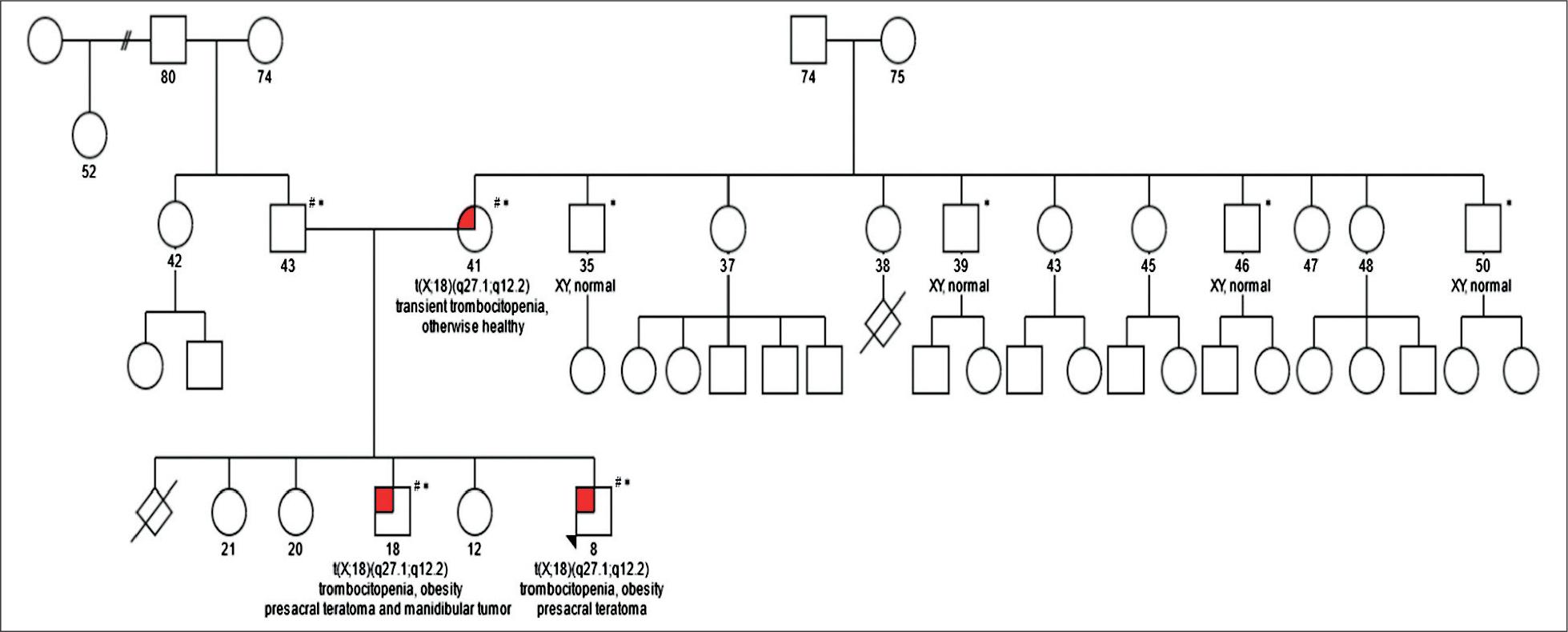
Family pedigree and segregation analysis results.
# family members in whom karyotyping, microarray and NGS were performed.
* family members in whom OGM was performed.
OGM requires a special isolation/extraction step, producing ultra-long/high molecular weight DNA (hm-wDNA) molecules, that are typically in the 200 kilobases (kb) to megabase (Mb) range, in contrast to typical DNA isolation protocols where the resulting DNA is usually up to 20 kb in size. Therefore, archival DNA samples cannot be used for OGM, and fresh extraction is needed. After extraction, DNA is labeled across specific motifs using the DLE-1 enzyme, while the backbone DNA is also labeled using a special stain. The current technical limitations of OGM concern the size of DNA required, DLE-1 labeling limitations, and interpretation challenges. As large DNA molecules are needed for this method, it currently cannot be performed from archived DNA or FFPE, and therefore fresh samples are needed. Furthermore, the method cannot detect SV within regions that do not contain the DLE-1 labeling motif, such as Robertsonian translocations. Similarly, regions spanning segmental duplications, e.g. on the chrY chromosome can result in several alternative assemblies. The interpretation of genomic variants in terms of pathogenicity is currently based on recommendations from ACMG and the joint consensus of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen)15,27. However, many of the different SVs detected by the OGM method, for example, balanced translocations, inversions, etc. lack clear guidelines for classification, and so the interpretation of these SVs needs to be carefully considered on a case-by-case basis. The limited size of known normal OGM genetic variation at the moment means, that many identified variants remain variants of unknown significance. Finally, because of its novelty, there is a need to establish a larger database of normal human genomic variation detected using OGM. When possible, the results should be confirmed by using an independent method, while for many rare disease cases, a trio setup is preferable to resolve causality.
Despite an extended segregation analysis, based on strictly applied ACMG criteria and ClinGen guidelines, the identified translocation remains a variant of unknown significance, highlighting the complexity of diagnostic results in rare disease cases as well as the remaining limitations of this technology. Hopefully, the future increase in healthy control population OGM variants and the establishment of official guidelines on the clinical interpretation of OGM variants will resolve many current interpretation challenges.
The diagnostic journey in case of rare disease is often complex and consist of many steps. In our case, the traditional karyotyping was negative and was followed by microarray. While microarray identified a duplication of 18q12.2 that was initially classified as a variant of unknown significance, after segregation showed it to be of maternal origin, it was reclassified as likely benign, and the patients were referred for exome sequencing. After initial exome sequencing was negative, reinterpretation with additional gene panels was performed in the proband, and following another negative result, was followed up by panel exome sequencing of over 2000 genes in trio setup, which also failed to identify causative variants, and the probands were referred for OGM. OGM showed a t(X;18) (q27.1;q12.2) translocation, that was confirmed to be of maternal origin and had the previously observed duplication as an accompanying event: ogm[GRCh38] t(X;18) (q27.1;q12.2)(140408784~140427850;38878133~3939 6298)mat,dup(18)(18q12.2)(38927193_39426970)mat. Despite extended segregation, we did not identify any additional healthy male carriers of the translocation in the family. Therefore, pending reinterpretation and possible functional assessments that may become possible in the future with additional technologies, the identified familial translocation currently remains a variant of unknown significance.
In conclusion, OGM represents a useful new method in the repertoire of genomic diagnostics available at CIGM UMCL, however applying ACMG criteria and ClinGen guidelines to SVs remains demanding, highlighting the complexity of the modern genomic diagnostic approach to rare disease testing. In our experience, currently, a major limitation of the method remains the difficulty of interpretation due to its novelty and the lack of healthy control population variants, which will hopefully be resolved and will increase the diagnostic yield of this method in the future. To circumvent this limitation, in the testing of rare disease patients, OGM in a trio setup is currently advised.