
Tuber himalayense B.C. Zhang & Minter belong to Ascomycota, Pezizomycetes, Pezizales, Tuberaceae, Tuber (Kirk et al. 2001; 2008). Tuber is predominantly distributed within temperate forest ecosystems across the northern and southern hemispheres. Europe, North America, and Southeast Asia are primary distributional regions (Mabru et al. 2004). Tuber is classified as an ectomycorrhizal fungus, engaging in symbiotic associations with various tree species belonging to Pinaceae, Fagaceae, Betulaceae, and Salicaceae (Trappe 1979). Notable genera include Pinus, Corylus, Cedrus, Picea, Abies, Larix, Tsuga, Pseudotsuga, Quercus, Fagus, Castanea, Betula, Carpinus, Populus, Salix, Tilia, Eucalyptus, Arctostaphylos, Arbutus, among others (Zanini et al. 1995; Chen 2003; Deng 2012). Tuber establishes symbiotic relationships with trees, enhancing soil structure, boosting soil fertility, and fostering plant growth, which positively affects the health and sustainable development of forest ecosystems. The colonization of Tuber can significantly increase the height of host plants, but the effects on the root-shoot ratio, stem diameter, and crown width of host plants are inconsistent (Plett et al. 2022; Xu et al. 2022). The rhizosphere provides the critical symbiocenosis for Tuber-host plant mutualism, wherein pedological parameters govern Tuber ontogeny (primordia differentiation, mycelial proliferation, sporocarp maturation). Calcareous rendzinas underlying Tuber niches exhibit optimal pedostructure: friable texture (pH 6.5–8.5), moderated hydric regimes, and humus-rich aerated horizons that potentiate ectomycorrhizal dialogues (Kües and Martin 2011). Tuber and host plants are mutually beneficial symbiosis, but also physical and metabolic interactions with harmful microorganisms, neutral microorganisms, and beneficial microbial communities in other niches (Frey-Klett et al. 2007; Bonfante and Anca 2009). These microorganisms include soil microorganisms, plant endophytic microorganisms, endophytic and epiphytic microorganisms of ectomycorrhizal fungi (Gryndler et al. 2013; Hashidoko et al. 2014; Splivallo et al. 2015; Benucci and Bonito 2016). Rhizosphere microorganisms play a crucial role in the formation of Tuber mycorrhiza (Thioulouse et al. 2012). They provide the necessary nutrients and conditions for the growth of Tuber mycorrhiza by decomposing organic matter, releasing nutrients, and producing hormones (Soudzilovskaia et al. 2015). At the same time, rhizosphere microorganisms can also affect the morphology and structure of mycorrhizas by interacting with Tuber mycorrhizas, thereby affecting their functions and effects. The relationship between rhizosphere microorganisms and Tuber mycorrhiza and host plants is not a simple one-way effect. There is a complex interaction network between them, and any change in microorganisms may impact the entire ecosystem. So far, no report has detailed the population characteristics of mycorrhizal microorganisms associated with T. himalayense. In this study, high-throughput sequencing technology was used to analyze the microbial community composition, diversity and other characteristics of rhizosphere soil samples of Tuber himalayense-Corylus heterophylla with different growth vigor. These were combined with the changes of soil physical and chemical properties to explore the community diversity and biochemical function of rhizosphere microorganisms and to provide a theoretical basis for better understanding the operation law of ecosystem and the construction of T. himalayense plantation.
Aiming at the demonstration base of mycorrhiza seedlings in Chanxi Town, Yinjiang County, Tongren City, Guizhou Province, China, we accurately measured the ground diameter and tree height of Tuber himalayense-Corylus heterophylla. According to their actual growth conditions, Tuber himalayense-Corylus heterophylla were categorized into three grades: excellent (CHTG), good (CHTM), and general (CHTB). Three trees were selected for growth index determination for each grade. To determine the ground diameter, a vernier caliper was employed to gauge the trunk’s thickness approximately 1cm above the soil level. The height from the base to the apex was measured using a tape measure.
Rhizosphere soil (0–20 cm depth) from Tuber himalayense-Corylus heterophylla plantations was collected using a standardized soil corer. Sampling design was as follows: for each plantation grade (n = 3 grades), three representative trees were selected, with five spatially independent sub-samples (1 m from trunk) per tree collected aseptically. Sub-samples from the same tree were homogenized into a single composite sample (total composite samples per grade: 3 trees × 1 composite/tree = 3 biological replicates). The total sample size was 9 (3 grades × 3 biological replicates). Samples were immediately stored in sterile bags on ice and processed within 4 hours. Each composite sample was split into aliquots: one stored at 4°C for physicochemical analysis; the other flash-frozen in liquid nitrogen and stored at –80°C for DNA extraction.
Total nitrogen (TN), total phosphorus (TP), total potassium (TK), available potassium available potassium (AK), available phosphorus (AP), alkaline hydrolysis nitrogen (AN), organic matter (OM), and pH were measured. Total nitrogen (TN) was determined by the Kjeldahl apparatus (Bremner 1996). Total phosphorus (TP) and total potassium (TK) were determined by sodium hydroxide fusion-molybdenum antimony anti-colorimetric method (Lu 2000) and flame photometric method (Bao 2000), respectively. Available potassium (AK) was determined by flame photometric method (Olsen and Sommers 1982). Available phosphorus (AP) was determined by molybdenum antimony anti-colorimetric method (Murphy and Riley 1962). Soil alkali-hydrolyzable nitrogen was assayed by alkali solution diffusion method (Lu 2000); organic matter (OM) was determined by potassium dichromate-sulfuric acid oxidation method (Walkley and Black 1934). Soil pH was measured by pH meter. Each sample was measured three times.
The collected soil samples were used to extract total soil DNA using the soil DNA extraction kit (SPINeasy DNA Kit for Soil) according to the instructions. The identification of fungi and bacteria was performed via metabarcoding using targeted amplicon sequencing. PCR protocols were as follows: bacterial 16S rRNA gene region (primers 515F/806R) and fungal ITS1 region (primers ITS1-F/ITS1-R) were amplified using KAPA HiFi HotStart (25 cycles, annealing at 55°C) and Phusion High-Fidelity (30 cycles, annealing at 56°C), respectively, with 0.25 μl of 20 mg/ml BSA added to suppress inhibitors. Amplified products were purified with AM-Pure XP beads and validated using an Agilent 2100 Bioanalyzer (Caporaso et al. 2011; Klindworth et al. 2013). This was followed by paired-end sequencing on the Illumina NovaSeq platform to construct small fragment libraries (16S V4: ~290 bp; ITS1: 250–400 bp).
Data preprocessing in MS Excel 2019 included IQR-based outlier removal and min-max normalization. Statistical analyses were performed in SPSS 25.0. Diversity metrics (Shannon/Chao1, Bray-Curtis) and LEfSe biomarkers were analyzed. Bacterial and fungal profiles were analyzed via BMKCloud using NovaSeq 6000, employing QIIME2/DADA2 for the bacterial ASV resolution and BLASTn/UNITE for fungal taxonomic annotation. SparCC networks visualized in Gephi identified modular communities. PICRUSt2-KEGG predictions were validated by Mantel tests.
Nine rhizosphere soil samples were collected. The CHTG sample originated from soil that supported robust growth of Tuber himalayense-Corylus heterophylla, exhibiting an average tree height of 117.33 cm and an average ground diameter of 1.47 cm. The CHTM sample was the soil with good growth of Tuber himalayense-Corylus heterophylla, with an average tree height of 80.67 cm and an average ground diameter of 1.40 cm. The CHTB sample was derived from soil supporting the general growth of Tuber himalayense-Corylus heterophylla, where trees attained an average tree height of 54.67 cm and an average ground diameter of 0.73 cm. The results (Fig. 1) showed that TP and AP in CHTG soil were superior to those in CHTM and CHTB, whereas TK was inferior to those in CHTM and CHTB. Conversely, OM, TN, AN, and pH were elevated in CHTM soil compared to CHTG and CHTB, with AK being lower. TK and AK in CHTB soil were more abundant than in CHTG and CHTB, while OM, TP, AN, AP, and pH were diminished. Despite the observed variations in nutrient content among the three rhizosphere soil groups, these differences were not statistically significant (p > 0.05).
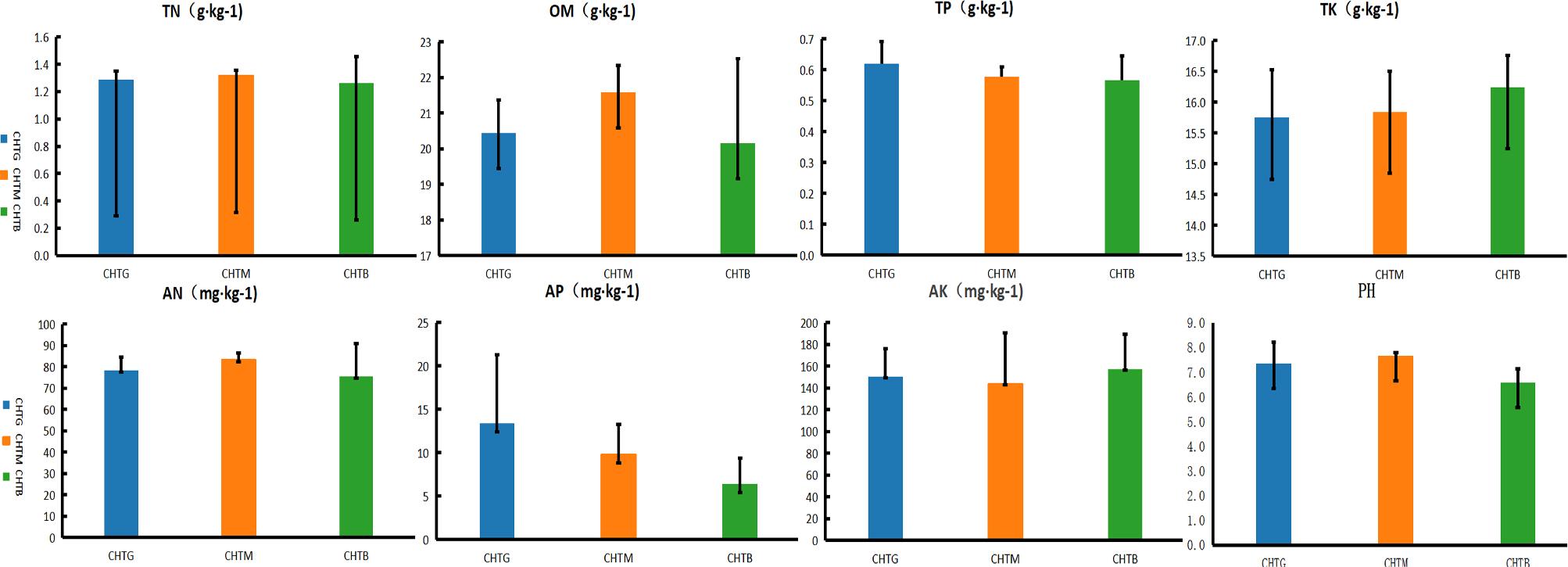
Soil nutrient content of three soil samples.
Nine samples were sequenced, with three replicates per sample, specifically for three rhizosphere soil samples. The fungal sequencing results showed that each sample’s sequencing number exceeded 70,000, and the dilution curve tended to the plateau period. After clustering, 1,814 valid OTUs (97% similarity) were obtained. The results of bacterial sequencing revealed that each sample’s sequencing count surpassed 70,000, and the dilution curve also approached the platform period (Fig. 2b), indicating suitability for further data analysis. After clustering, 8,344 valid OTUs (97% similarity) were obtained.
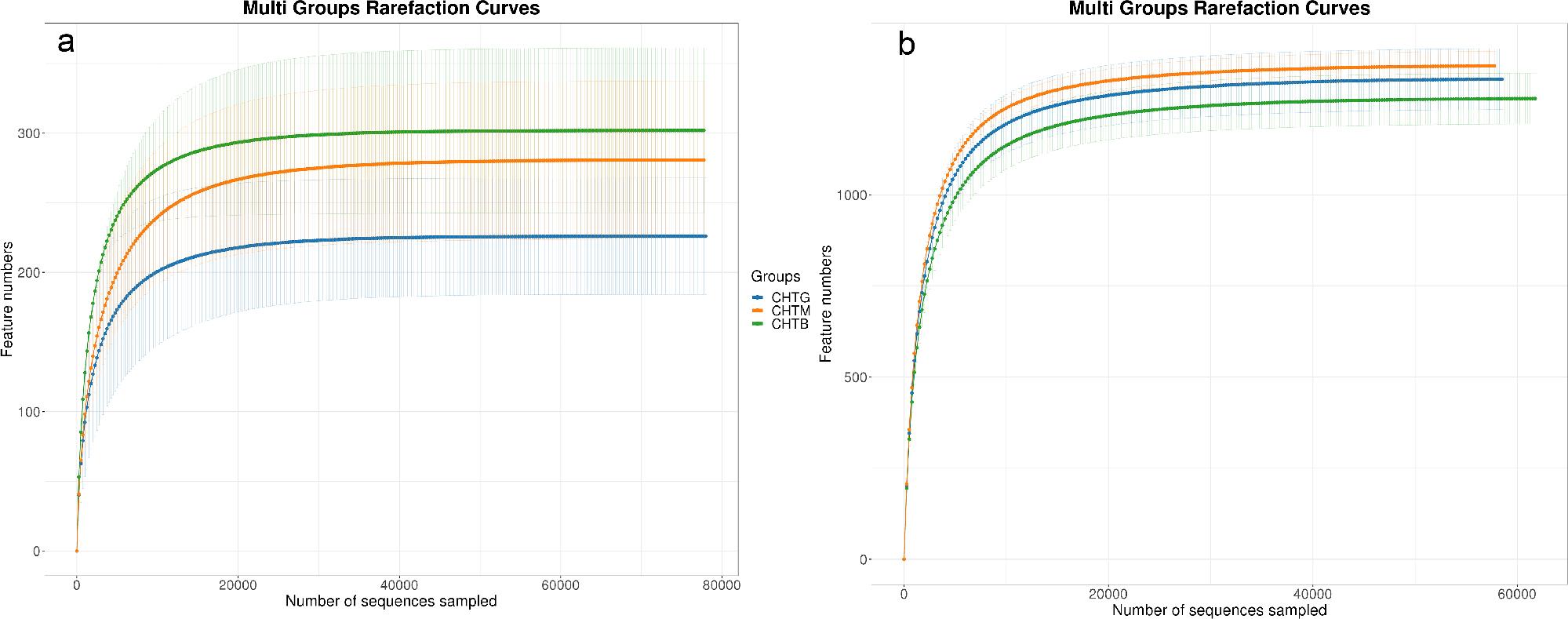
Samples rarefaction Curve.
The Chao1, Shannon and Simpson index of fungi in CHTG were lower than those in CHTM and CHTB (Fig. 3), suggesting that the fungal community in CHTG exhibits reduced species richness and diversity compared to CHTM and CHTB. Conversely, the Chao1, Shannon and Simpson index of bacteria in CHTG were comparable to those of CHTM and CHTB (Fig. 3), indicating that the bacterial communities in CHTG, CHTM, and CHTB possess similar levels of species richness and diversity.
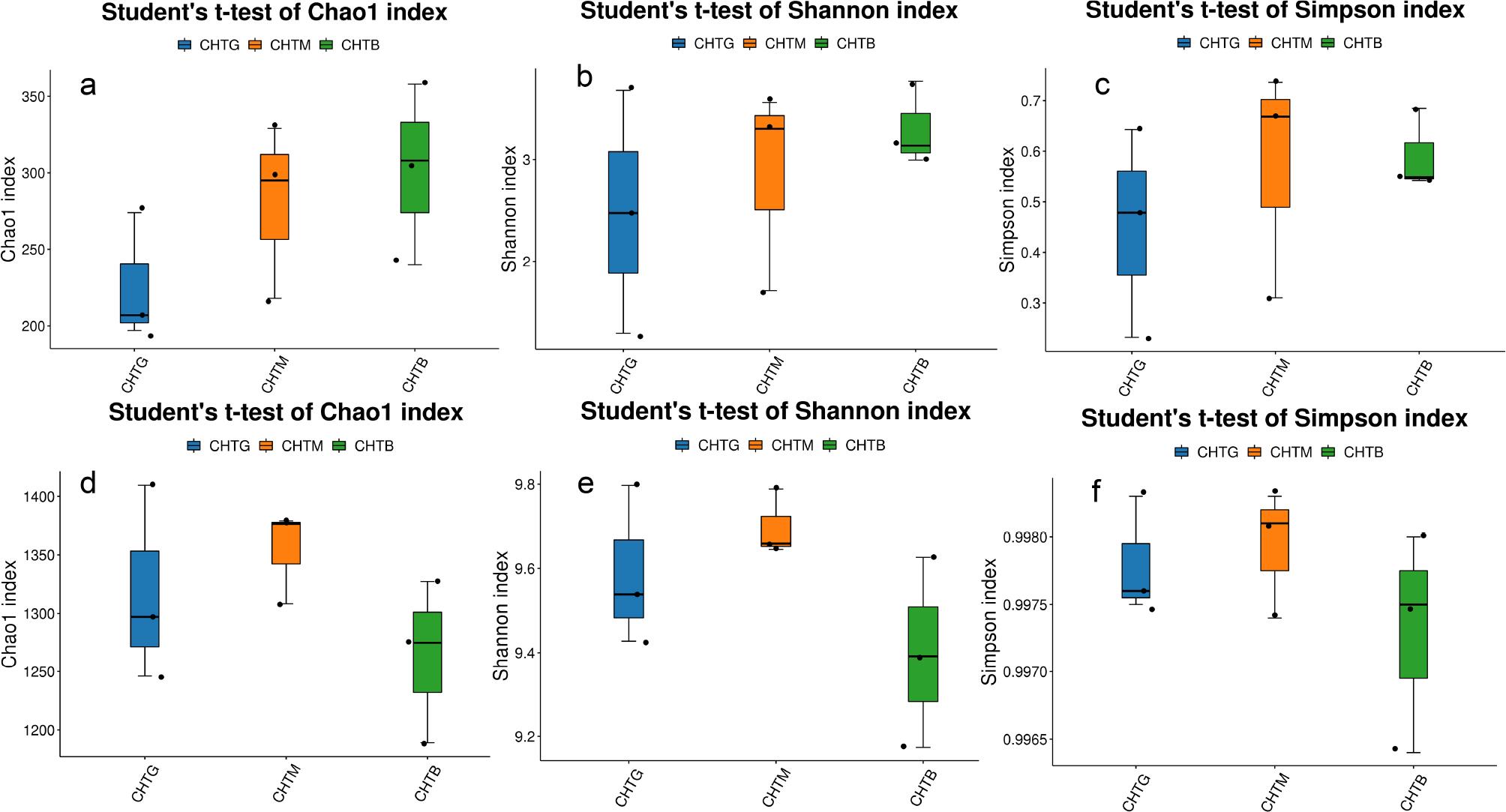
Soil microbial diversity index of fungi (a, b, c) and bacteria (d, e, f).
The principal component analysis (PCA) results at the microbial genus level (Fig. 4) showed that the composition of rhizosphere bacteria and fungi in the three groups of samples was different, consistent with the results of the Venn diagram. Notably, the CHTG and CHTM groups exhibited a closer distance, suggesting a higher similarity in their rhizosphere microbial communities’ species and quantity composition. The comparison between CHTG and CHTM did not present a significant disparity, suggesting a close correlation between tree growth status and the structure of their rhizosphere microbial communities. The microbial communities associated with the rhizosphere of both well-growing and medium-growing trees appeared relatively stable and exhibited minimal differences. In contrast, the microbial community structure of badly growing trees in or around the rhizosphere may have experienced substantial alterations. Such changes could be attributed to adverse growth conditions, including soil nutrient deficiencies, water scarcity, pollution, etc. These detrimental environmental factors might impede the growth and proliferation of beneficial microorganisms while fostering the proliferation of pathogens, ultimately leading to restricted tree growth.
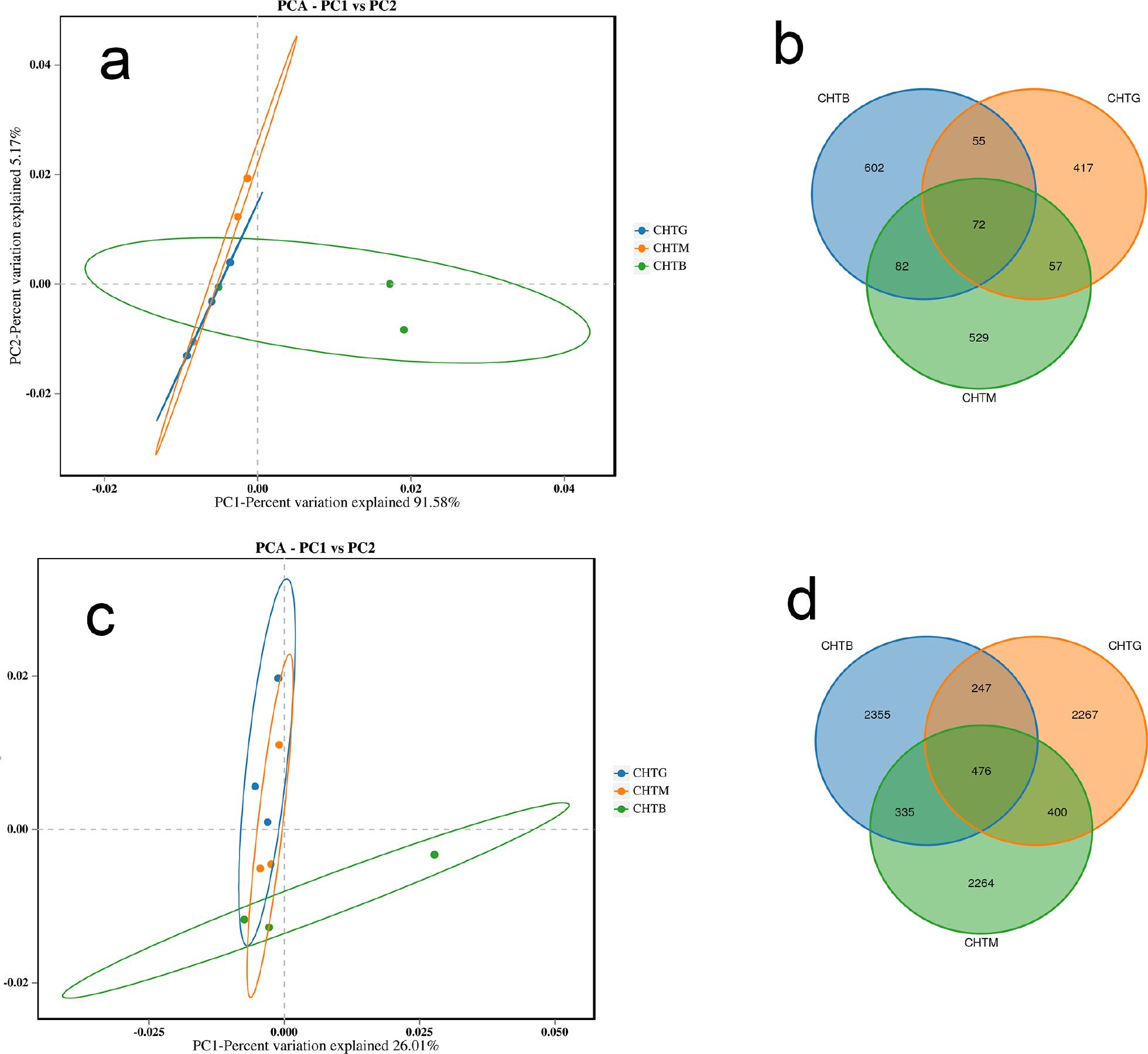
Comparative analysis of fungal and bacterial communities through Principal Component Analysis (PCA) and OTU-level Venn diagrams: fungal communities (a, b); bacterial communities (c, d).
High-throughput sequencing technology was used to investigate the diversity of soil microorganisms in CHTG, CHTM, and CHTB. These three bacterial groups were predominantly categorized into 30 phyla, and the relative abundance of the dominant bacterial groups at the phylum level was used to construct a species accumulation diagram (Fig. 5a). The dominant bacteria in the soil were Proteobacteria, Acidobacteriota, and Actinobacteriota. The relative abundance of Acidobacteriota in CHTG increased by 7.84% and 8.31% compared with CHTM and CHTB. The relative abundance of Proteobacteria in CHTM soil increased by 0.87% and 12.87% compared with CHTG and CHTB soil. The relative abundance of Actinobacteriota in CHTB soil increased by 0.44% and 1.89% compared with CHTM and CHTB soil. Despite the similarity in the dominant phylum across the three soil bacterial communities, variations in relative abundance were observed. Further analysis of the relative abundance of the samples at the family level showed that compared with CHTM and CHTB (Fig. 5b), the dominant families in CHTG were still the dominant families in CHTM and CHTB soils, but the relative abundance changed. The relative abundance of Nitrosomonadaceae increased by 6.4% and 30.26%, respectively, and the relative abundance of Vicinamibacteraceae increased by 27.09% and 137.55%, respectively. The relative abundance of Sphingomonadaceae increased by 38.04% and 118.27%, the relative abundance of Comamonadaceae increased by 5.80% and 119.94%, and the relative abundance of Xanthobacteraceae decreased by 10.07% and 18.47%, respectively. These results indicate that the community structure of the three soil bacteria in this study has undergone significant alterations. The dominant phylum remains consistent at the level of the three soil fungi, although the relative abundance varies. In Fig. 5c, the dominant fungi in the soil mainly included Ascomycota and Basidiomycota. At the phylum level, the main phylum composition of the three soil fungi was similar. At the level of fungal family (Fig. 5d), Tuberaceae was the highest at the soil fungal community structure level. Compared with CHTM and CHTB, the relative abundance of Tuberaceae in CHTG increased by 17.87% and 15.58%, respectively. The second and third dominant families were different. CHTG were Cladosporiaceae and Thelephoraceae, CHTM were Hydnangiaceae and Thelephoraceae, and CHTB were Cladosporiaceae and Mycosphaerellaceae. In this study, the community structure of the three soil fungi was similar at the phylum level, and the difference was significant at the family level. The relative abundance changed significantly, indicating the complexity and diversity of the soil fungal community. This difference may be due to the growth status of different mycorrhizal seedlings and their adaptability to the soil environment.
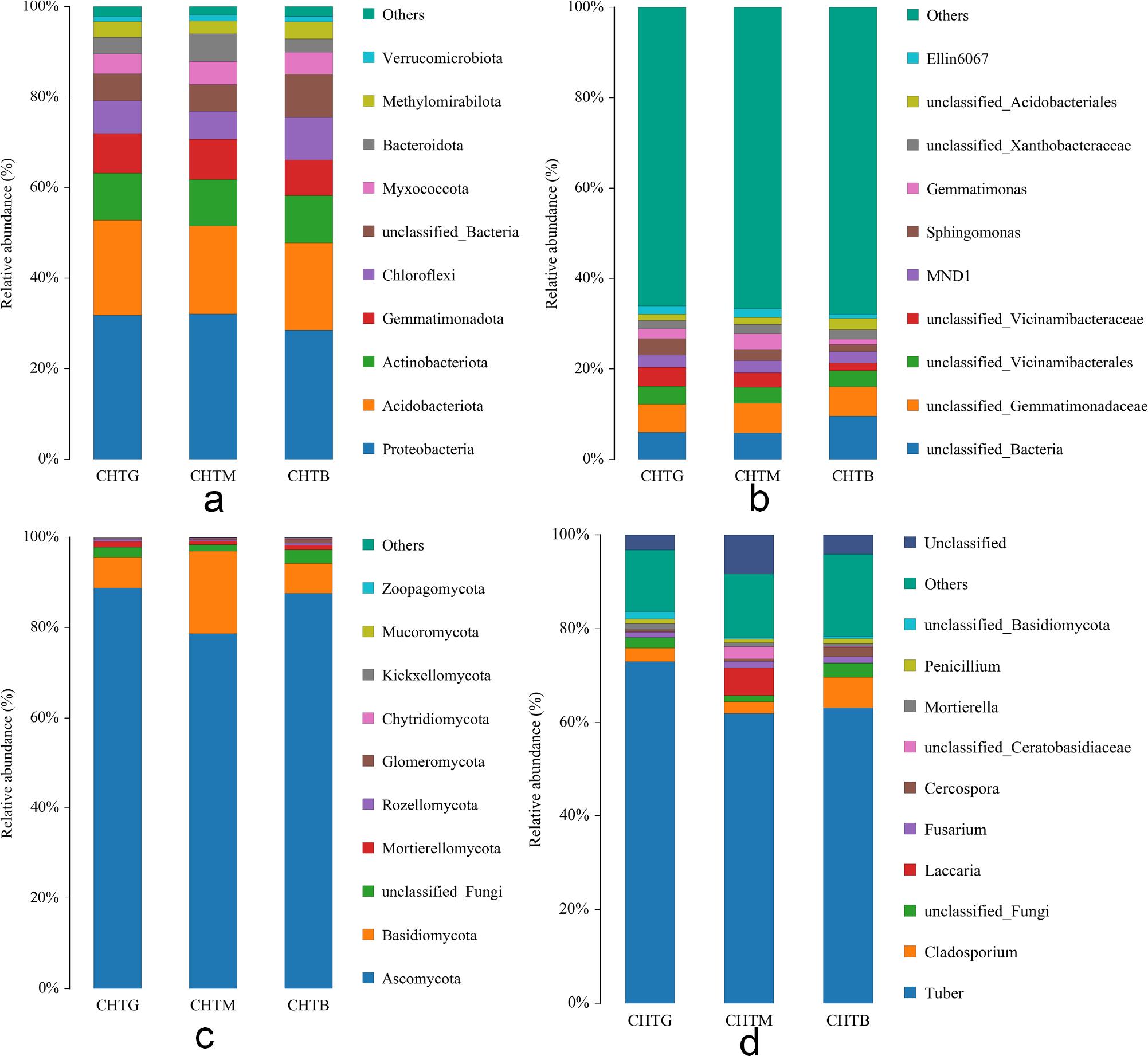
Taxonomic profiling of rhizosphere microbial communities: Bacterial composition at phylum and family levels (a, b); Fungal composition at phylum and genus levels (c, d).
Mycorrhizal seedlings that are thriving may possess a more robust nutrient absorption capacity and environmental adaptability, which could facilitate the proliferation of Tuberaceae, allowing it to become the dominant family within the community. Cladosporiaceae and Thelephoraceae were found in both good-growing and bad-growing mycorrhizal seedlings, yet their relative abundances varied, which may reflect the universality and adaptability of these families in soil. Conversely, Hydnangiaceae and Mycosphaerellaceae only have certain advantages in good and general growth of mycorrhizal seedlings, respectively, which may mean that these families are more sensitive to specific conditions of the soil environment. By examining the family-level variations within the soil fungal community structure, we can delve deeper into how these distinctions influence soil functionality and the growth of mycorrhizal seedlings. For instance, a rise in the abundance of Tuberaceae may enhance soil fertility and improve soil structure, thereby creating more favorable conditions for the growth of mycorrhizal seedlings.
Concurrently, the competition and symbiotic relationships among various fungal families can significantly influence the stability and functionality of soil ecosystems. Therefore, through an in-depth study of the composition and structure of soil fungal communities, we can better understand the function and stability of soil ecosystems and provide a scientific basis for agricultural production and ecological environment protection. Furthermore, it aids in developing innovative soil enhancement techniques and biological control strategies to boost crop yields and quality, thereby fostering the sustainable development of agriculture.
Based on the RDA/CCA analysis of the selected main soil nutrient factors, Fig. 6 shows that the abundance of MND1 is positively correlated with all the main soil nutrient factors, indicating that MND1 are conducive to the absorption of M, P, K and other nutrients by Tuber himalayense-Corylus heterophylla. The two bacterial groups of Ellin6067 and Gemmatimonas exhibit a positive correlation with TN, AN, OM, TP, AP and pH, which may enhance the absorption of organic matter, N, and P elements by Tuber himalayense-Corylus heterophylla. The population of Sphingomonas was positively correlated with AP and TP, indicating that this group can transform the phosphorus that was difficult to be directly used by plants into an effective form by decomposing organic matter and releasing phosphatase, thereby increasing the phosphorus supply in the soil and promoting the growth of Tuber himalayense-Corylus heterophylla. Tuber emerged as the dominant genus, demonstrating positive correlations with soil pH but negative correlations with AN, OM, TN, TP, TK, AK, and AP, consistent with Zeng et al. (2022) and Gryndler et al. (2013). As the dominant genus, Tuber was positively correlated with soil pH, and negatively correlated with AN, OM, TN, TP, TK, AK, and AP. The negative correlation of TK was the most pronounced, indicating that Tuber favors slightly alkaline soils and that their growth is physiologically constrained in environments with high potassium levels. Tuber was negatively correlated with other highly abundant genera, such as Cladosporium, Penicillium, and Mortierella. The strongest negative correlation was observed with Penicillium, indicating that the distribution of Tuber and Penicillium in soil may be limited by similar resources or environmental conditions, leading to a mutually exclusive ecological relationship. This relationship could be attributed to competition for nutrients, water, or other living conditions, or it may be due to inhibitory effects from their respective metabolites.
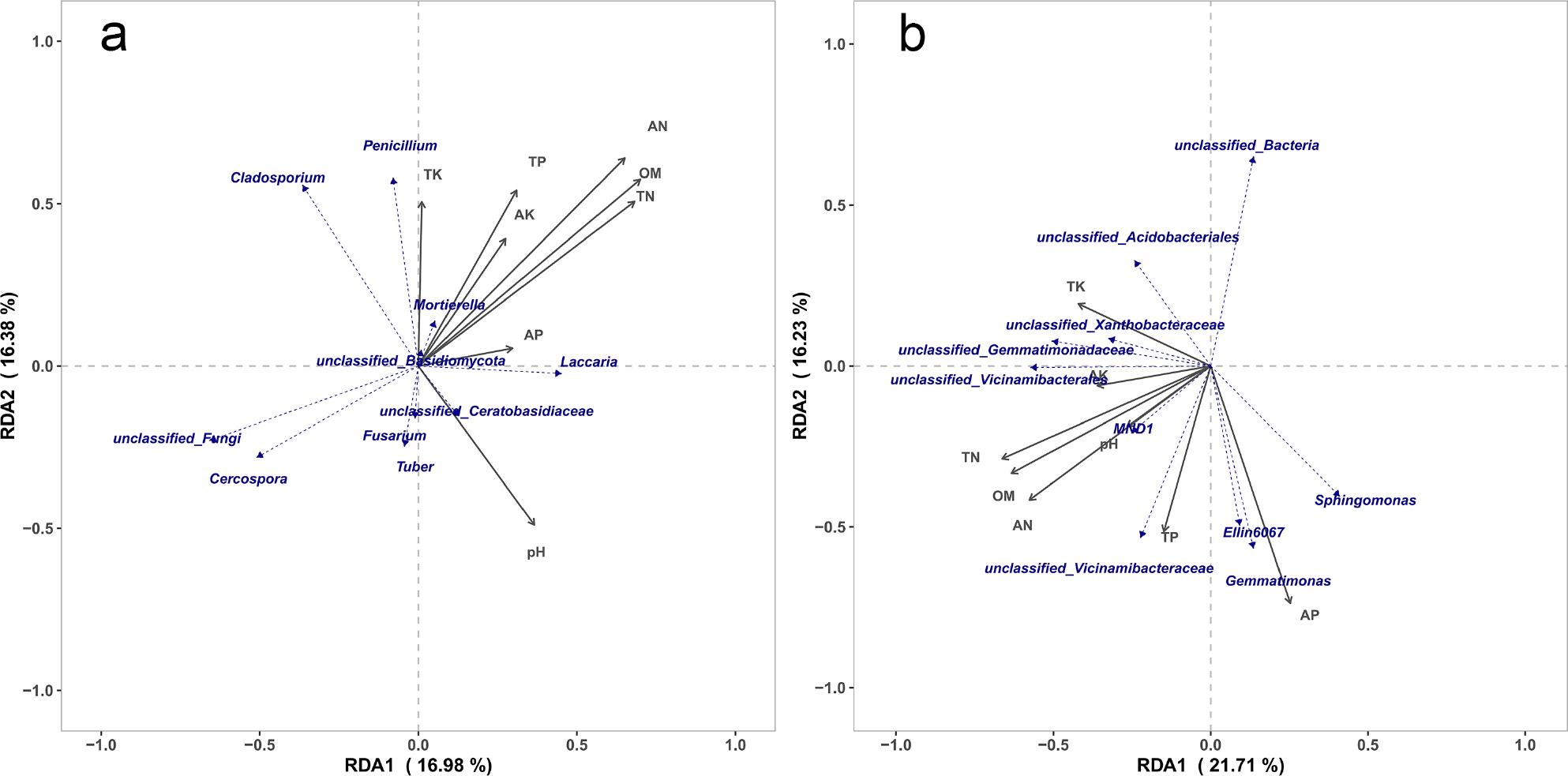
Redundancy analysis (RDA) of fungi (a) and bacteria (b) community structure and environmental factors.
Symbiotic networks effectively show interactions and changes in microorganisms. They are widely used to analyze changes in microbial communities. Table I shows that bacterial modularity and clustering coefficients are lower than those of random networks, while fungal ones are higher. This suggests bacteria have uniform node connectivity without prominent clusters, whereas fungi have distinct clusters and a modular, ‘small world’ network structure. The topological parameters of bacteria and fungi in these networks are notably different. The clustering coefficient, graph density, graph diameter, average path length, average degree, betweenness centrality, and degree centrality of bacteria were higher than those of fungi, while the modularity of bacteria is notably lower at 0.463. The extensive modularity of fungi (0.527) indicates that the symbiotic network structure between bacteria and fungi exhibits significant differences. These differences not only reflect the different niches and functions that bacteria and fungi occupy within the ecosystem, but they may also have a crucial impact on the stability and functionality of the ecosystem. Therefore, conducting an in-depth investigation into the symbiotic network structure of bacteria and fungi, along with its determinants, is paramount for comprehending microbial ecosystems’ functionality and stability.
Symbiotic network topological parameters.
| Network Parameters | Fungal | Bacterial |
|---|---|---|
| Nodes | 46 | 42 |
| Edges | 78 | 100 |
| Clustering coefficient | 0.364 | 0.607 |
| Graph density | 0.075 | 0.116 |
| Graph diameter | 18.615 | 24.27 |
| Average path length | 3.565 | 4.663 |
| Average degree | 3.391 | 4.762 |
| Betweenness centralization | 0.170 | 0.322 |
| Degree centralization | 0.146 | 0.177 |
| Modularity | 0.527 | 0.463 |
Employing the SPIEC-EASI framework (Kurtz et al. 2015), we constructed genus-level microbial co-occurrence networks to investigate symbiotic dynamics between T. himalayense and Corylus heterophylla across ecological gradients. These networks integrate the top 50 bacterial and fungal genera by relative abundance, systematically revealing taxon-specific interaction patterns under varying edaphic conditions (Fig. 7). In this network, each circle symbolizes a species, with its size corresponding to the average abundance of that species. The lines connecting the circles denote the correlation between pairs of species, where the line thickness indicates the intensity of the correlation. The line’s color is as follows: red signifies a positive correlation, green indicates a negative correlation. In the bacterial network diagram (Fig. 7a), the average species richness of unclassified bacteria was observed to be the highest, exhibiting a positive correlation with Acidibacter and a negative correlation with Gemmatimonas and Elin6067, albeit weakly. The two dominant genera with strong correlation were uncultured Firmicutes bacteria and uncultured Proteobacteria, which were positively correlated. This may imply that the environment may harbor a highly diverse bacterial community, where these unclassified bacteria could interact with various environmental factors, potentially influencing the stability of the entire ecosystem. In the fungal network diagram (Fig. 7b), Tuber exhibited the highest average species richness, which was negatively correlated with Fusarium and unclassified Sordariomycetes. The two most dominant genera, unclassified Didymellaceae and Plectosphaerella, displayed a strong positive correlation. This suggests that Tuber himalayense-Corylus heterophylla has established a unique and stable ecosystem, featuring a mutual exclusion mechanism with Fusarium, unclassified Sordariomycetes, and other fungi. In the two network diagrams, a positive correlation was observed among most genera of bacteria and fungi, with the dominant genera exhibiting strong correlations. This suggests synergistic or symbiotic relationships between these microbial communities, which may collectively enhance the stability and development of the ecosystem. For instance, the positive correlation between uncultured Firmicutes bacteria and uncultured proteobacterium could imply their involvement in specific biochemical reactions or metabolic processes, thereby supporting the ecosystem’s material cycling and energy conversion. Similarly, the positive correlation between unclassified Didymellaceae and Plectosphaerella may also mean that they perform analogous functions and roles in specific ecological processes.
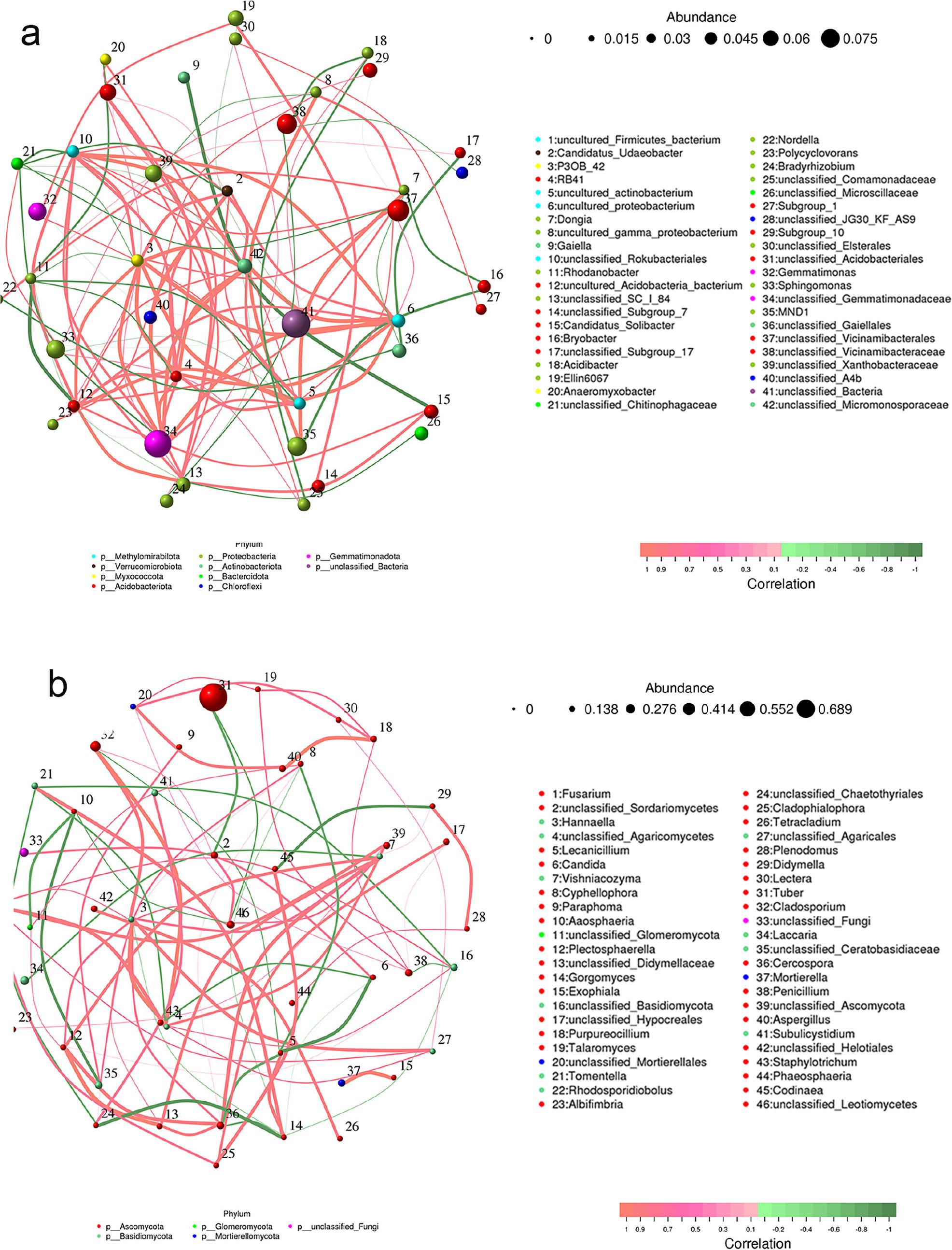
Topological profiling of bacterial (a) and fungal (b) symbiont networks.
The circle represents the species, and the size of the circle represents the average abundance of the species; line represents the correlation between the two species, the thickness of the line represents the strength of the correlation, the color of the line: red represents a positive correlation, green represents a negative correlation.
Phylogenetic functional prediction was performed using PICRUSt2 v2.4.1 (Douglas et al. 2020) through the following workflow: 16S rRNA sequences were phylogenetically placed against the IMG database (v.5.0) using SATé-enabled phylogenetic placement (Chen et al. 2019); Gene content was inferred from nearest sequenced taxa (≥ 97% identity) with hidden-state prediction algorithms (Parks et al. 2014); KEGG Orthology (KO) abundances were mapped to MetaCyc metabolic pathways using MinPath parsimony (Kanehisa et al. 2016). Combined with Fig. 8, it can be seen that the three with the highest relative abundance percentage of metabolic pathways in this prediction results are: global and overview maps, carbohydrate metabolism, amino acid metabolism. At the bacterial classification level, the dominant bacteria are mainly Bacteroidota, Chitinophagia, Chitinophagales, and Chitinophagaceae. Fig. 9 shows that fungal functional gene predictions are categorized by nutritional methods into three groups, with pathotrophs having the highest relative abundance, followed by saprotrophs and symbiotrophs. Cercospora has the greatest abundance, followed by Codinaea and Laccaria. This shows that the soil samples of Tuber himalayense-Corylus heterophylla have a complex microbial community structure, potentially involving a broad spectrum of interactions and symbiotic relationships among these microorganisms. Key species Cercospora, Codinaea, and Laccaria drive community interactions through differentiated ecological strategies: Cercospora, as a facultative saprophyte-weak pathogen, utilizes carbon sources (organic acids/polysaccharides) from mycorrhizal exudates and benefits from truffle metabolites suppressing competitors; Codinaea acquires host photosynthates via mycorrhizal networks while secreting chitinases for antipathogen collaboration; Laccaria, a known ECM symbiont, may facilitate nutrient exchange (e.g., nitrogen, phosphorus) and root architecture modulation, potentially competing with Tuber for host carbon allocation. This tripartite interplay underscores a finely balanced trophic network where saprotrophic decomposition, pathogenic pressure, and mutualistic symbiosis co-regulate soil nutrient cycling and ecosystem resilience. Such complexity highlights the need to evaluate functional redundancy and niche partitioning among these guilds to optimize truffle cultivation and soil health management.
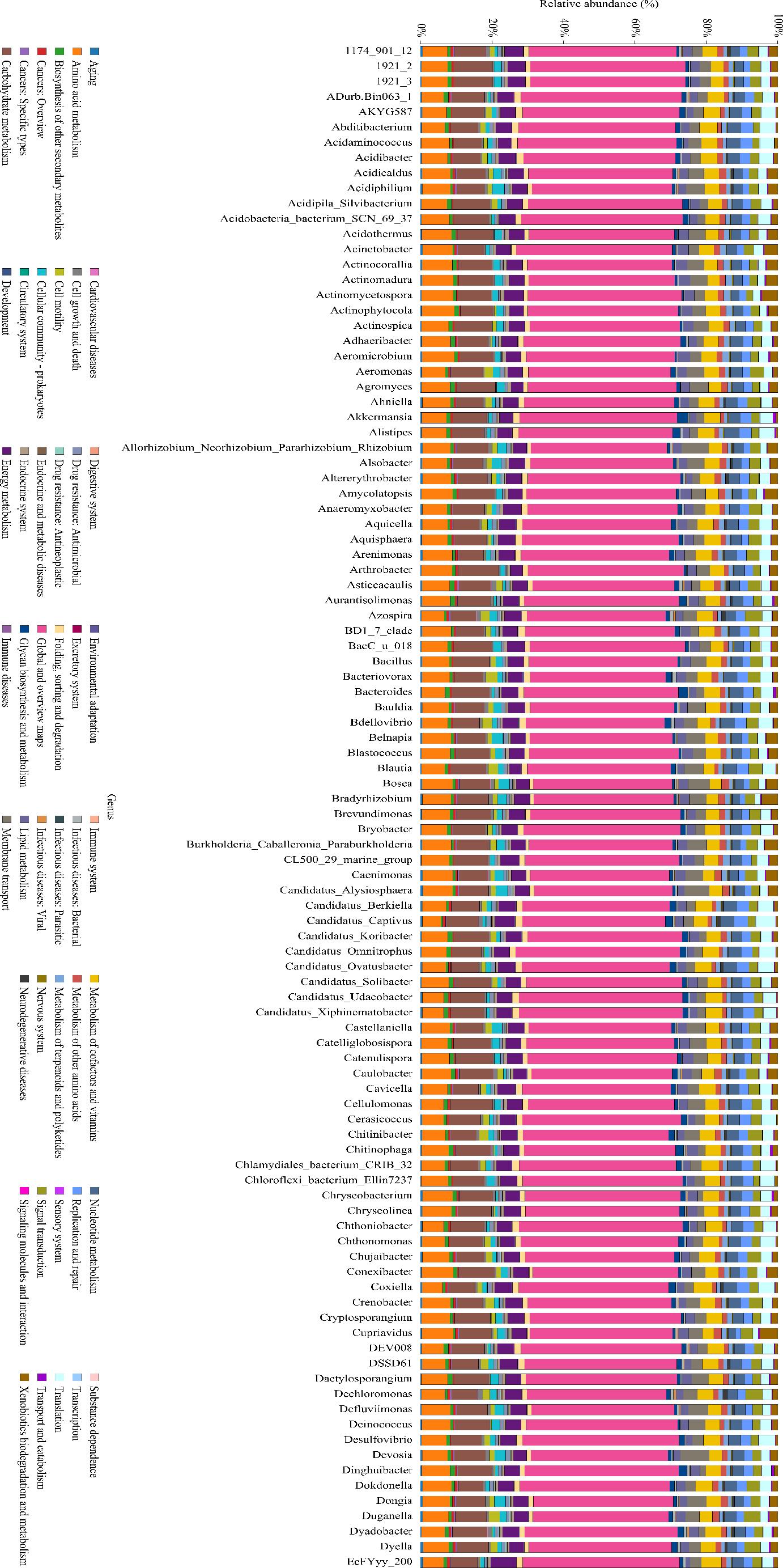
KEGG metabolic pathway histogram.
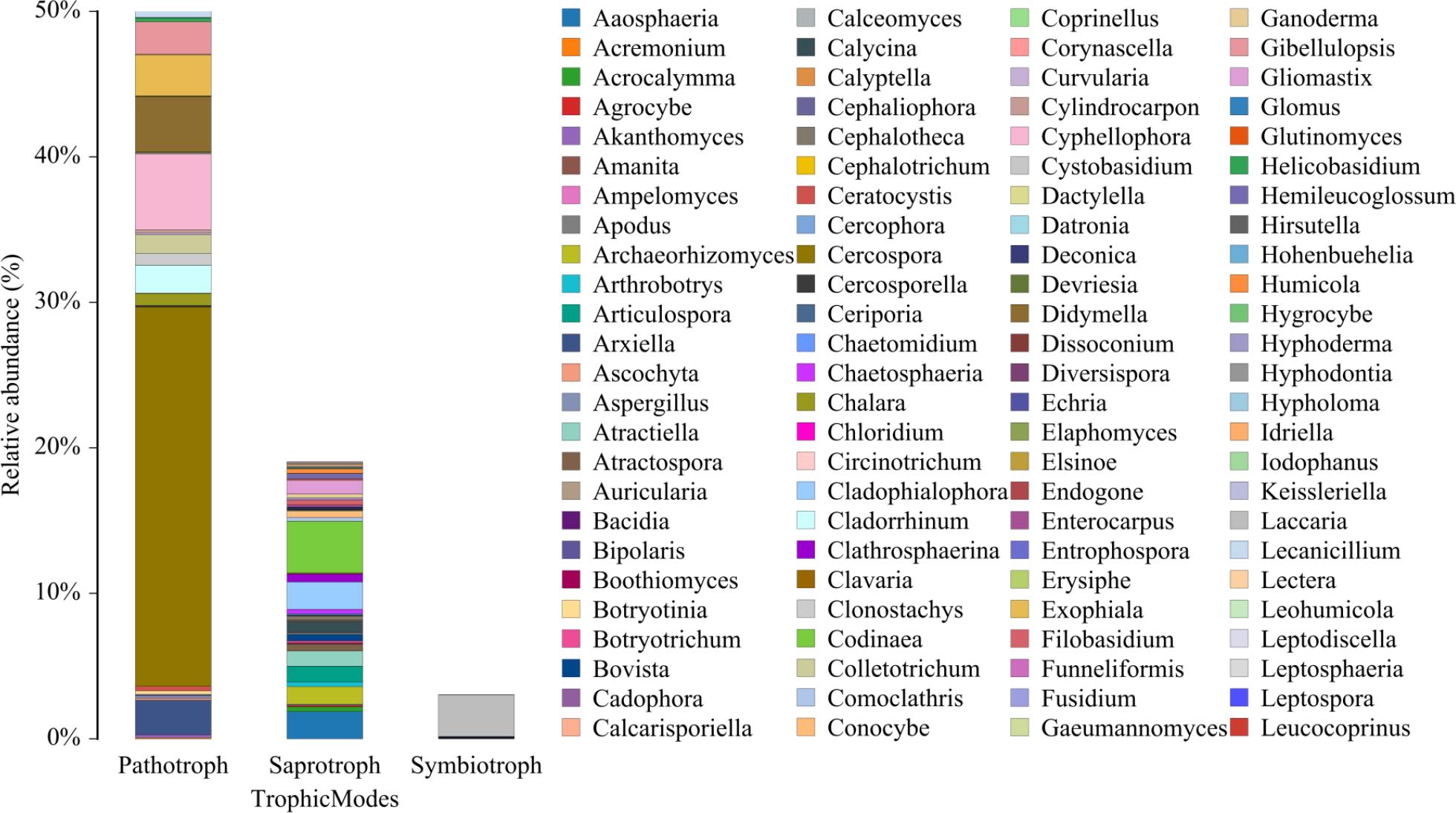
Funguild taxonomic composition bar plot.
In this study, the Illumina NovaSeq sequencing platform was used to sequence the 16S and ITS regions of soil microorganisms in the rhizosphere of different mycorrhizal seedlings. The results showed that the dominant bacteria were Proteobacteria, Acidobacteriota, and Actinobacteriota, among which Proteobacteria was the most abundant, accounting for about 30.80% of the total sequence, which was consistent with the reports of Wan et al. (2015) and Deng et al. (2018). The dominant fungal phyla were Ascomycota and Basidiomycota, of which Ascomycota was the most abundant, accounting for about 84.33% of the total sequence. It indicated that the microbial community in the rhizosphere soil of truffle mycorrhizal seedlings with different growth vigor and types had a relatively stable structure. Further analysis showed some differences in microbial communities in the rhizosphere soil of mycorrhizal seedlings with different growth vigor. The relative abundance of Acidobacteriota in the rhizosphere soil of good-growing mycorrhizal seedlings was higher than that of medium-growing and bad-growing seedlings. Combined with the analysis of soil physical and chemical properties, these bacteria may play an important role in the absorption of P source, pH adjustment, and maintenance of soil structure (Fierer et al. 2007; Jones et al. 2009; Lauber et al. 2009; Ward et al. 2009; Navarrete et al. 2013). The relative abundance of Ascomycota in the rhizosphere soil of good-growing mycorrhizal seedlings was higher than that of medium-growing and bad-growing mycorrhizal seedlings, indicating that mycorrhizal colonization had a good effect on the growth of hazelnut (Rincón et al. 2005; Domínguez Núñez et al. 2008). Therefore, we conclude that Tuber colonization enhances soil organic carbon stabilization and nutrient flux optimization in the truffle-hazelnut ecosystem by recruiting stress-resilient microbial consortia (e.g., Acidobacteriota for phosphorus cycling, Ascomycota for symbiosis), while suppressing competitors through metabolite-mediated interactions. This tripartite synergy underpins host vigor and agroecosystem sustainability.
The physicochemical properties and microbial diversity of rhizosphere soil showed that the Chao1 index, Shannon and Simpson index of fungi in CHTG soil were lower than those in CHTM and CHTB. In addition, the principal component analysis at the genus level and the species composition analysis at the species level found that the three soil bacteria were similar at the dominant phylum level. However, the differences at the family level were large. Moreover, there were significant differences between bacteria and fungi in the symbiotic network under different growth conditions of hazelnut mycorrhizal seedlings. The average richness of unclassified bacterial species in the bacterial network was the largest, which was positively correlated with Acidibacter and negatively correlated with Gemmatimonas and Elin6067. Studies have shown that the dominant factors for the formation of soluble inorganic nitrogen SIN and soluble organic nitrogen SON by Acidibacter (Zhong et al. 2018) promote nitrification to form ammonia and small molecule nitrogen, which has a significant effect on the colonization of truffles and the activity of roots (Kang et al. 2020; 2022). In the fungal network, the average richness of Tuber species was the largest, which was negatively correlated with Fusarium and unclassified Sordariomycetes. The first two dominant genera with strong correlation were unclassified Didymellaceae and Plectosphaerella, respectively, indicating that Tuber species were complementary to unclassified Didymellaceae and Plectosphaerella in ecological functions. Alternatively, they share some key resources in the habitat. Tuber, as an important mycorrhizal fungus, can form a symbiotic relationship with plant roots and promote the absorption of nutrients by plants. Plectosphaerella and Unclassified Didymellaceae may play an important role in decomposing organic matter and releasing nutrients, providing an essential nutrient source for the growth and development of Tuber.
Dominant flora plays a crucial role in determining the balance of microbial communities. It influences ecosystem functions through higher abundance and dominant species with important energy transformation, and can affect the composition and structure of microbial communities (Li et al. 2024). At the phylum level, the rhizosphere soil fungi of the three Tuber himalayense-Corylus heterophylla were mainly Ascomycota, followed by Basidiomycota. In the study of rhizosphere soil microbial diversity of truffle hazelnut mycorrhizal seedlings, it was found that most of the Ascomycota were symbiotroph, indicating that the mycorrhizal seedlings had constructed a stable mycorrhizal system and occupied a significant dominant position in the ecosystem. Basidiomycota primarily function as saprophytes, secreting an array of extracellular enzymes that facilitate degradation, such as lignin enzymes, which can decompose lignin and other difficult-to-degrade substances (Bardgett and Mcalister 1999; Bossuyt et al. 2001), promoting soil nutrient cycling and improving soil quality (Sui et al. 2016). The bacteria in the rhizosphere soil of the three Tuber himalayense-Corylus heterophylla were mainly Proteobacteria, followed by Acidobacteriota, Actinobacteriota. This bacterial community composition aligns with patterns observed in other Tuber-host systems, including T. indicum (Li et al. 2017; Deng et al. 2018; Zeng et al. 2022), T. aestivum (Benucci and Bonito 2016), T. melanosporum (Antony-Babu et al. 2014; Deveau et al. 2016), and T. borchii (Li et al. 2019). The soil types, Tuber and symbiotic host plants taken in the above studies are inconsistent, but the dominant bacterial groups in the mycorrhizal soil are similar. Proteobacteria, Acidobacteriota, and Actinobacteriota are the dominant bacterial groups in the mycorrhizal soil. At the genus level, Tuber emerged as the dominant genus among the soil fungi associated with Tuber himalayense-Corylus heterophylla soil fungi, indicating that regardless of the growth of Tuber himalayense-Corylus heterophylla after transplanting, Tuber has formed a unique and stable ecosystem. Unclassified Bacteria is the common dominant genus of bacteria in the rhizosphere soil of three Tuber himalayense-Corylus heterophylla. It may be because most of the bacteria are uncultured, have no complete genomic information, have not been named, and have many deficiencies in the database. In addition, some species with lower abundance may play a potentially important role in maintaining the stability of the microbial ecosystem of mycorrhizal seedlings, which is a potential factor for studying the microecological stability of the rhizosphere soil of Tuber mycorrhizal seedlings. For example, Gemmatimonas are the dominant genus in the rhizosphere of Suillus luteus, which has certain ecological functions such as promoting the absorption of mineral elements by plant roots, inhibiting pathogens, and promoting the development of rhizosphere soil micro-ecological environment in a healthy direction (Luo et al. 2021).
The in-situ detection of rhizosphere soil nutrients and microbial diversity in various mycorrhizal seedlings has revealed that the stable mycorrhizal system remains intact following the transplantation of Tuber himalayense-Corylus heterophylla. However, the abundance of Tuber significantly impacts the growth of these mycorrhizal seedlings. Rhizosphere fungi and bacterial communities of Tuber himalayense-Corylus heterophylla, exhibiting varying growth vigor, are similar in their principal groups, with a greater richness and diversity. Functional gene prediction indicates a synergistic or symbiotic relationship between bacteria and fungi. Most fungi are pathotrophs, with strong lignin decomposition and carbon fixation capabilities. The bacterial community mainly focused on key metabolic processes, such as carbohydrate and amino acid metabolism, and jointly promoted the stability and development of the ecosystem. The results of this study laid the foundation for the mycorrhizal synthesis of Tuber, the fine management of plantations, the ecosystem carbon sink role, and the investigation of rhizosphere soil microbial function, thus clarifying the direction for future research.