
Invasive pulmonary aspergillosis (IPA) is a highly lethal infectious disease caused by Aspergillus that invades the bronchus and lung tissue (Baddley et al. 2010). IPA is caused by the interaction between inhaled Aspergillus spores and host immune effector cells and usually occurs in patients with impaired immune function. Neutropenia is a typical risk factor for the disease. In addition, the incidence of IPA has been increasing in immunocompromised patients who are not neutropenic but are undergoing glucocorticoid therapy, receiving solid organ transplants, admitted to intensive care units, or suffering from acquired immunodeficiency syndrome and chronic respiratory diseases (Kosmidis and Denning 2015). It is highly clinically significant to study the mechanism of immune response in IPA and to explore new diagnostic and treatment methods for IPA from the perspective of altering the host immune state.
The human gut harbors many microorganisms involved in maintaining host immune homeostasis. The gut microbiota may play a role in pulmonary infection by expressing pathogen-associated molecular patterns, activating immune cells, regulating cytokine levels, and producing metabolites, such as short-chain fatty acids (SCFAs), which reach the lungs through the lymphatic or circulatory system (Villena and Kitazawa 2020). Pulmonary infections can alter the richness and composition of gut microbiota, weakening the respiratory tract’s immune defense and promoting disease development. Distinctive changes in the gut microbiota of diseased populations have been observed in bacterial pneumonia caused by Streptococcus pneumoniae (Schuijt et al. 2016) and Klebsiella pneumoniae (Wu et al. 2020) and viral pneumonia caused by coronaviruses (Shen et al. 2022), influenza viruses, and respiratory syncytial viruses (Groves et al. 2018). These changes have also been found to be correlated with disease susceptibility and prognosis.
In pulmonary fungal infections, gut microbiota can induce the production of immune cells such as regulatory T cells (Tregs) and T helper 17 cells, contributing to anti-fungal immune responses (Scheffold and Bacher 2020). Additionally, oral administration of bacteria that express elevated levels of alpha-Gal has been shown to help prevent pulmonary aspergillosis (Mateos-Hernández et al. 2020). Animal models of IPA have demonstrated decreased diversity and altered gut microbiota composition, leading to an imbalance in intestinal homeostasis, possibly affecting intestinal immune tolerance and promoting inflammatory responses (Kulas et al. 2019). However, there is limited understanding regarding potential alterations in the composition and functionality of the gut microbiota in patients with IPA. Hérivaux et al. (2022) showed that changes in the pulmonary microbiota of patients with IPA could predict the prognosis of the disease; nevertheless, the impact of gut microbiota on the prognosis of IPA remains unreported in the literature.
This study aimed to analyze the composition and diversity of the gut microbiota in patients with IPA using metagenomic next-generation sequencing (mNGS) technology. Additionally, we aimed to identify the microbial species and functional pathways that may be related to IPA, explore the relevant mechanisms, and identify potential biomarkers that can be used to diagnose or predict the prognosis of the disease to provide new insights for prevention and immune intervention.
This study included 43 patients diagnosed with IPA (IPA group) at the Department of Respiratory and Critical Care Medicine of the Third Affiliated Hospital of Soochow University and 31 healthy participants (H group) from the physical examination center between October 2021 and October 2023. Fecal samples for mNGS were collected from the participants.
The inclusion criteria for patients with IPA were as follows: i) age ≥ 18 years; and ii) “proven” and “probable” cases of IPA meeting the diagnostic criteria of the European Organization for Research and Treatment of Cancer and the Mycoses Study Group (Donnelly et al. 2020).
The exclusion criteria for patients with IPA were as follows: i) suffering from digestive tract tumors, infection, and inflammatory bowel disease; ii) suffering from active rheumatic immune system-related diseases; and iii) using oral probiotics or antibiotics and antifungal drugs within 1 month.
Laboratory test results and prognostic information of patients with IPA were collected from the hospital’s electronic medical records system. All patients included in the study or their families signed informed consent forms, and the study was approved by the Ethics Committee of our hospital (Section No. 075, 2023).
After the patients naturally defecated in a dry and clean bedpan, 1–2 g of feces was collected and placed in a DNA sample storage tube, which was then stored in a –80°C ultra-low temperature freezer until further testing.
All samples were collected according to standard procedures. According to the manufacturer’s instructions, DNA was extracted using the TIANamp Micro DNA Kit (TIANGEN Biotech(Beijing)Co., Ltd., China). The quantity and quality of the DNA were assessed using Qubit™ (Invitrogen™, Thermo Fisher Scientific Inc., USA) and NanoDrop™ (Thermo Scientific™, Thermo Fisher Scientific Inc., USA) instruments, respectively.
Following the manufacturer’s protocol, DNA libraries were prepared using the Hieff NGS™ C130P2 OnePot II DNA Library Prep Kit for MGI (Yeasen Biotechnology, China). The libraries were qualified using Agilent 2,100 and 50 bp single-end sequenced on MGISEQ-200 (MGI Tech Co., Ltd., China).
An in-house bioinformatics pipeline was used for pathogen identification. Initially, raw sequencing data was processed by splitting it with fastq v0.23.2 (Chen 2023). High-quality sequencing data was generated by removing low-quality reads, adapter contamination, duplicate, and short reads (less than 36 bp in length). Human host sequences were identified by mapping to the human reference genome (hs37d5) using the bowtie2 software v2.2.6 (Langmead et al. 2009). Reads that could not be mapped to the human genome were retained and aligned to the microorganism genome database for pathogen identification. Microbial taxonomic identification was conducted utilizing Kraken v2.0.7, while species abundance estimation was performed with Bracken v2.5.0. Our microorganism genome database was derived from sequences obtained from the NCBI database (https://www.ncbi.nlm.nih.gov), which were then processed through our in-house developed bioinformatics pipeline, resulting in a comprehensive library encompassing more than 20,000 species of bacteria, fungi, viruses, and parasites.
Functional metagenome profiling was performed using the HUMAnN2 standard workflow with the default settings (Franzosa et al. 2018). HUMAnN2 software was used to align the sequences of the microorganisms with those in the MetaCyc database (Caspi et al. 2020), annotate gene functions and metabolic pathways, and calculate the relative abundance of each pathway. The table of MetaCyc pathway abundance in units of reads per kilobase (RPK) was normalized to the relative abundance using the humann2_renorm_table command in HU-MAnN2. Finally, the species profiles and the MetaCyc pathway were retained for further analysis if they had a non-zero abundance in at least 40% of the samples.
Statistical analyses and figures were generated using R software v4.3.1 (R Core Team 2023). Shannon and inverse Simpson indices were calculated to evaluate the alpha diversity of microbial communities. Beta diversity was assessed using the Bray–Curtis measure and visualized using a principal coordinate analysis (PCoA) plot and principal component analysis (PCA). PERMANOVA was performed using the R package “vegan” to analyze the Bray–Curtis distance in the IPA and H groups. The Wilcoxon rank-sum test was used to assess differences in alpha diversity and the relative abundance of MetaCyc pathways, while the ANOVA-like differential expression-2 analysis was employed to identify differences in the relative abundance of microorganisms between the two groups. The filtration conditions for the microorganisms were as follows: sample detection rate > 10% and average abundance > 0.1%. Spearman’s correlations between the relative abundances of species level and MetaCyc pathways or clinical features were calculated using the R package “cor.test”. The random forest prediction model was constructed using species relative abundance as a random variable and all samples as the training set. The model includes multiple decision trees, each trained using randomly selected samples and features. Using the feature importance assessment method built into random forests, we calculated the mean decrease in accuracy and mean decrease in the Gini of species. The top 10 biomarkers with the highest contributions were selected based on feature importance scores. Furthermore, a new classifier model was constructed using the top five important biomarkers as classification labels to verify the model’s effectiveness. This study employed a two-tailed test method, considering a p-value below 0.05 indicative of significant differences between the groups.
This study included 43 patients (7 confirmed and 36 clinically diagnosed) diagnosed with IPA (IPA group) and 31 healthy examinees (H group). The demographic characteristics, clinical indicators, and underlying diseases are presented in Table I.
Comparison of general data between the two groups of patients.
| Project | IPA group (n = 43) | H group (n = 31) | p-value |
|---|---|---|---|
| Basic information | |||
| Sex (Male) [n (%)] | 32 (74.4) | 18 (58.1) | p>0.05 |
| Age (years) (IQR) | 73 (67 ~ 77) | 63 (54 ~ 82) | p>0.05 |
| Underline illness [n (%)] | |||
| Diabetes | 2 (4.7) | 1 (3.2) | p>0.05 |
| Hypertension | 11 (25.6) | 3 (9.7) | p>0.05 |
| Chronic obstructive pulmonary disease | 7 (16.3) | 0 | p>0.05 |
| Malignant diseases of the blood system | 7 (16.3) | 0 | p>0.05 |
| Rheumatic diseases | 5 (11.6) | 0 | p>0.05 |
| Solid organ malignancy | 2 (4.7) | 0 | p>0.05 |
| No underline diseases | 9 (20.9) | 27 (87.1) | p< 0.05 |
| Clinical indicators | |||
| White blood cell (109/l) | 6.3 ± 3.5 | – | |
| Neutrophil count (109/l) | 5.0 ± 3.1 | – | |
| Neutrophil percentage (%) | 73.9 ± 15.4 | – | |
| Eosinophil count (109/l) | 0.1 ± 0.1 | – | |
| Eosinophil percentage (%) | 1.5 ± 3.0 | – | |
| Lymphocyte count (109/l) | 0.9 ± 0.6 | – | |
| Lymphocyte percentage (%) | 17.5 ±11.8 | – | |
| Serum (1-3)-β-d-glucan (pg/ml) | 48.8 ±79.9 | – | |
| Serum galactomannan (μg/l) | 0.8 ± 1.3 | – | |
| Bronchoalveolar lavage fluid Galactomannan (μg/l) | 3.7 ± 4.0 | – | |
| Carcinoembryonic antigen (ng/ml) | 2.9 ± 2.6 | – | |
| C-reactive protein (mg/l) | 73.6 ±71.1 | – | |
| Procalcitonin (ng/ml) | 2.3 ± 5.9 | – | |
| APACHE II score | 16.1 ±10.0 | – | |
IPA group – invasive pulmonary aspergillosis; H group – healthy group; APACHE II score – Acute Physiology and Chronic Health Evaluation II score
To investigate changes in the gut microbiota of patients with IPA, we performed mNGS on 74 fecal samples obtained from patients with IPA (n = 43) and healthy individuals (n = 31). Initially, we used the Shannon and inverse Simpson indices to calculate the alpha diversity index of the enrolled individuals and thoroughly evaluated the species richness and evenness in each sample. Both methods showed that the alpha diversity of the gut microbiota was significantly lower in patients with IPA than in healthy controls (Fig. 1A). We then used the Bray–Curtis distance to assess the beta diversity of the microbiota and analyze whether there are differences in microbial composition between patients with IPA and healthy controls. The Bray–Curtis distance was visualized using PCoA and PCA. The results showed that, although the gut microbiota composition of patients with IPA overlapped with that of healthy controls, there was a general trend of separation and a statistical difference (Fig. 1B).
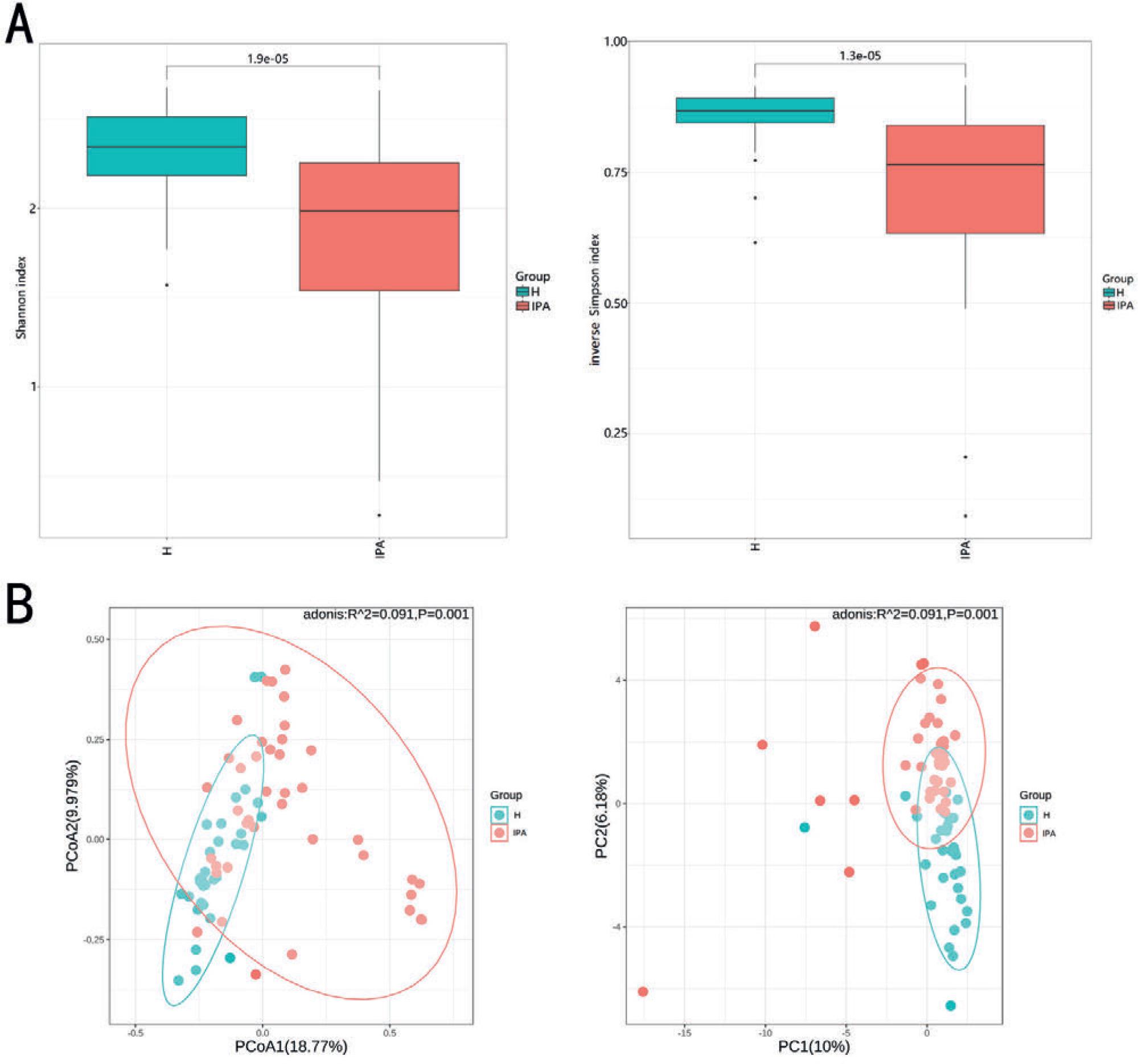
Comparison of alpha and beta diversity of gut microbiota between the H and IPA groups.
A)Alpha diversity analysis: Shannon index (p = 1.9 × 10−5), inverse Simpson index (p = 1.3 × 10−5); B) Beta diversity analysis: principal coordinate analysis (p = 0.001) and principal component analysis (p = 0.001) based on Bray–Curtis distance. H – healthy controls, IPA – invasive pulmonary aspergillosis.
We selected microorganisms with higher relative abundance rankings from the entire cohort at the phylum, genus, and species levels and analyzed the microorganisms with changes in the gut microbiota in patients with IPA compared to that in healthy individuals. First, we selected the top 11 microorganisms at the phylum level and found that Actinomycetota, Ascomycota, and Cossaviricota were significantly enriched in patients with IPA, whereas Bacteroidota and Bacillota were enriched considerably in healthy individuals (Fig. 2A). We then selected the top 20 microorganisms in terms of relative abundance in the entire cohort at both the genus and species levels. At the genus level, 15 genera were differentially enriched between the two groups, among which Hoylesella, Haemophilus, Finegoldia, Enterococcus, Escherichia, Enterocloster, Corynebacterium, and Staphylococcus were highly enriched in patients with IPA. However, Phocaeicola, Bacteroides, Blautia, Faecalibacterium, Mediterraneibacter, Roseburia, and Fusicatenibacter were significantly enriched in healthy individuals (Fig. 2B). At the species level, 8 bacterial species showed differential enrichment between the two groups. Prevotella disiens, Finegoldia magna, Enterococcus faecium, Escherichia coli, Enterocloster bolteae, and Corynebacterium pyruviciproducens were significantly enriched in patients with IPA. In contrast Blautia wexlerae, and Ruminococcus gnavus were significantly enriched in healthy individuals (Fig. 2C).
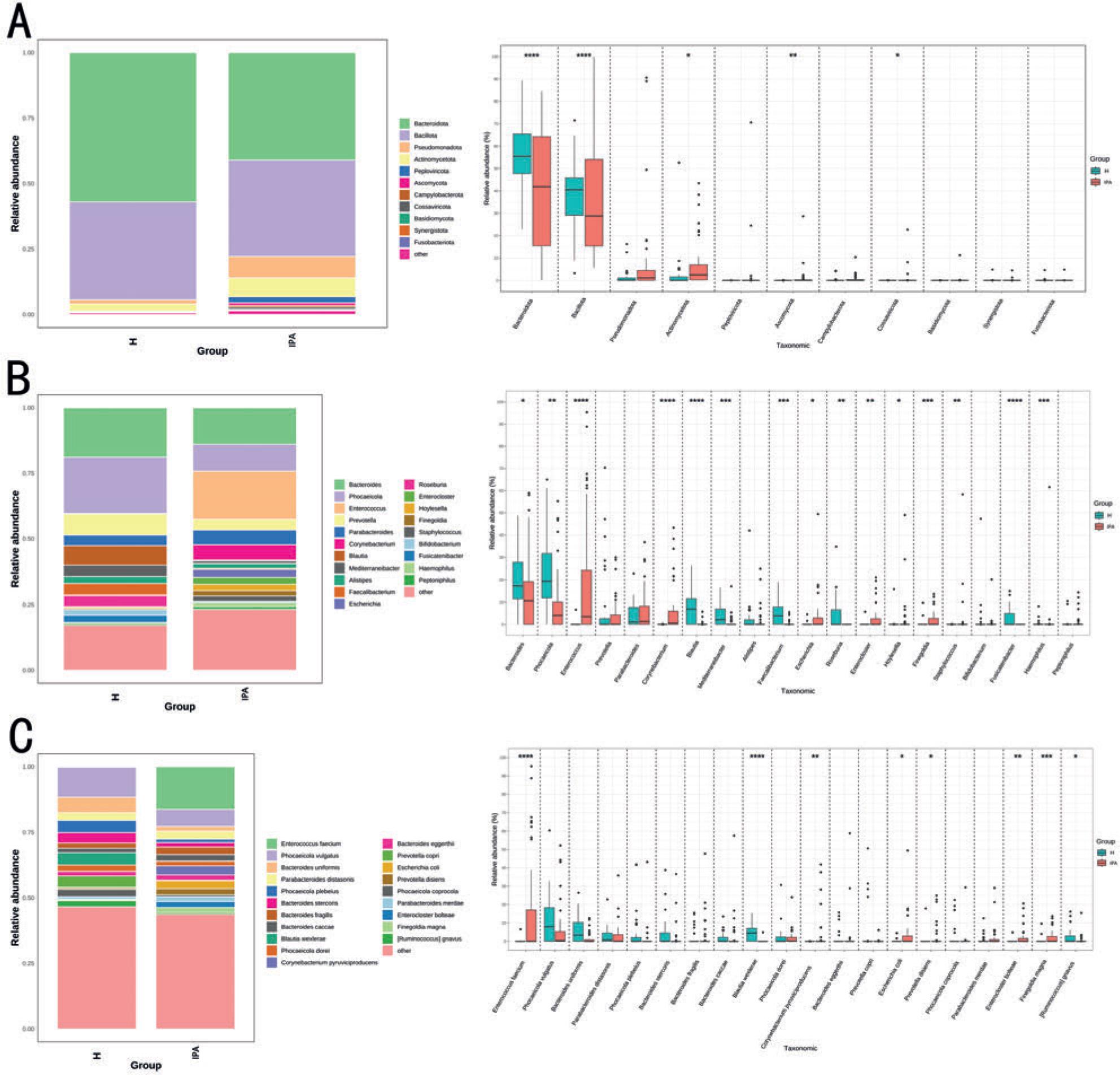
Microbial composition and different species analysis of the H and IPA groups at different levels of phylum, genus, and species.
A)Phylum-level microbial composition and differential species analysis; B) genus-level microbial composition and differential species analysis; C) species-level microbial composition and differential species analysis. (* p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001). H – healthy controls, IPA – invasive pulmonary aspergillosis.
The correlation between gut microbiota and gene function may imply that species mediate in the relevant pathways. Therefore, we selected the top 20 metabolic pathways in the H and IPA groups, analyzed their differences, and analyzed their associations with bacteria at the species level. Our results showed that these pathways were significantly enriched in the H group, suggesting that the overall metabolic level of the gut microbiota in patients with IPA is lower than that in healthy individuals. We found that E. faecium, Phocaeicola vulgatus, and B. wexlerae were associated with most metabolic pathways. Among them, metabolic pathways related to carbohydrate synthesis and degradation, pyruvate fermentation, CDP-diacylglycerol biosynthesis, and various amino acid, nucleoside, and nucleotide biosynthesis were positively correlated with B. wexlerae and P. vulgatus and negatively correlated with E. faecium. Therefore, we inferred that these three species play an important role in the metabolomics of the gut microbiota. Interestingly, we found that P. vulgatus, Phocaeicola coprocola, and Phocaeicola dorei were positively correlated with CDP-diacylglycerol biosynthesis. In addition, several species of Phocaeicola and Bacteroides were positively correlated with multiple metabolic pathways involved in nucleoside and nucleotide biosynthesis. The remaining significant correlations are shown in the heat map in Fig. 3.
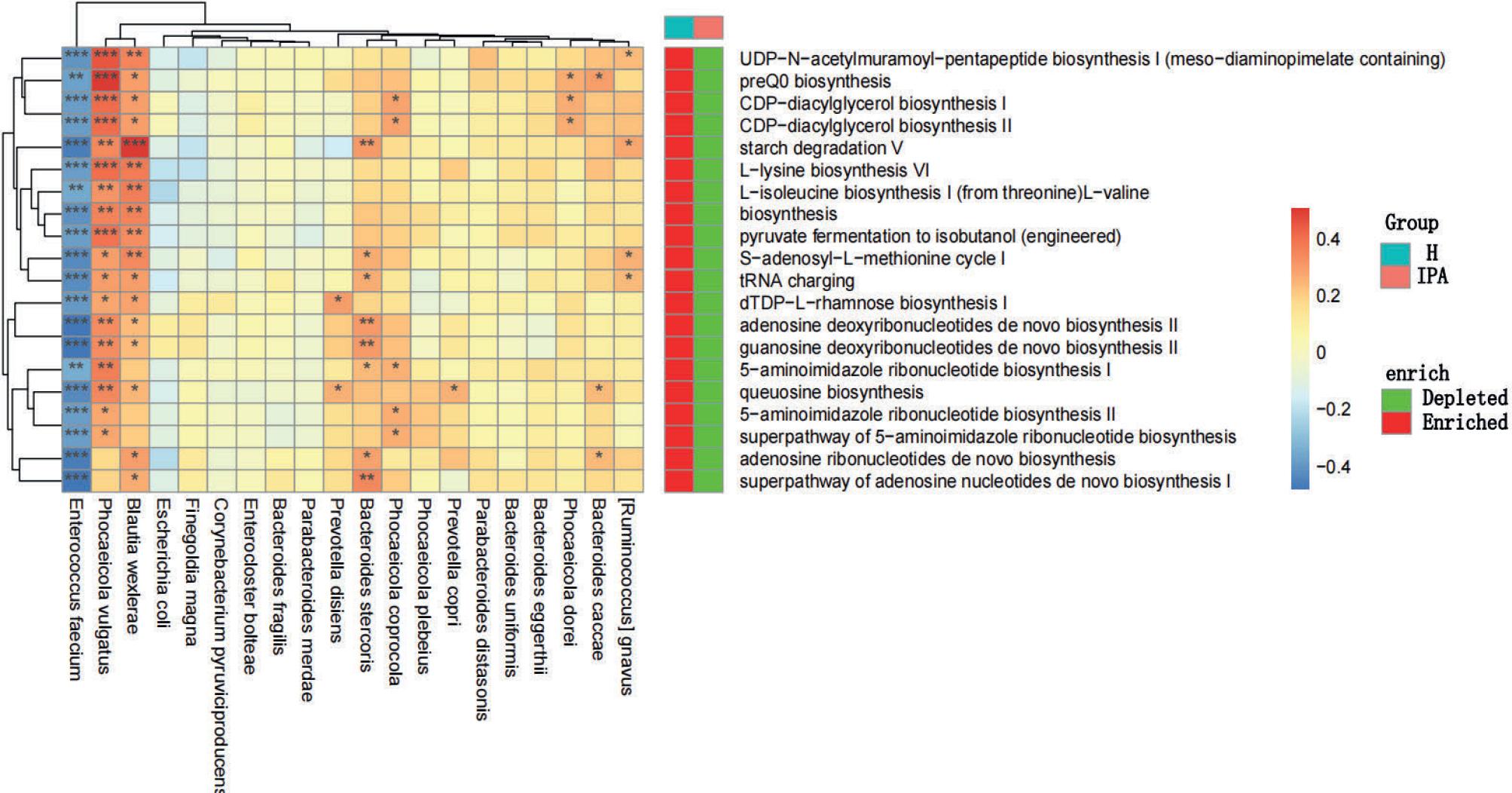
Differential analysis of metabolic pathways of the gut microbiota between the IPA and H groups and Spearman’s correlation analysis between metabolic pathways and gut microbiota.
The color depth in the heat map indicates the strength of the correlation: red indicates a positive correlation, while blue indicates a negative correlation (* p < 0.05; ** p < 0.01; *** p < 0.001). “Depleted” indicates that the metabolic pathway abundance is significantly reduced, and “Enriched” indicates that the metabolic pathway abundance is significantly increased (p < 0.05). H – healthy controls, IPA – invasive pulmonary aspergillosis.
Next, we selected the top 20 species regarding gut microbiota abundance among patients with IPA and further analyzed the correlation between these species and clinical indicators. The Acute Physiology and Chronic Health Evaluation II (APACHE II) scores were negatively correlated with Bacteroides uniformis and P. disiens. Bronchoalveolar lavage fluid galactomannan was negatively correlated with P. disiens and C. pyruviciproducens but positively correlated with Bacteroides thetaiotaomicron. Furthermore, Alistipes onderdonkii positively correlated with C-reactive protein and procalcitonin (PCT), whereas P. disiens and Hoylesella timonensis negatively correlated with PCT. In addition, lymphocyte count negatively correlated with Enterococcus faecalis and positively correlated with P. disiens. Neutrophil count positively correlated with E. faecium, and the neutrophil percentage negatively correlated with P. disiens. The remaining significant correlations are shown in the heat map in Fig. 4.
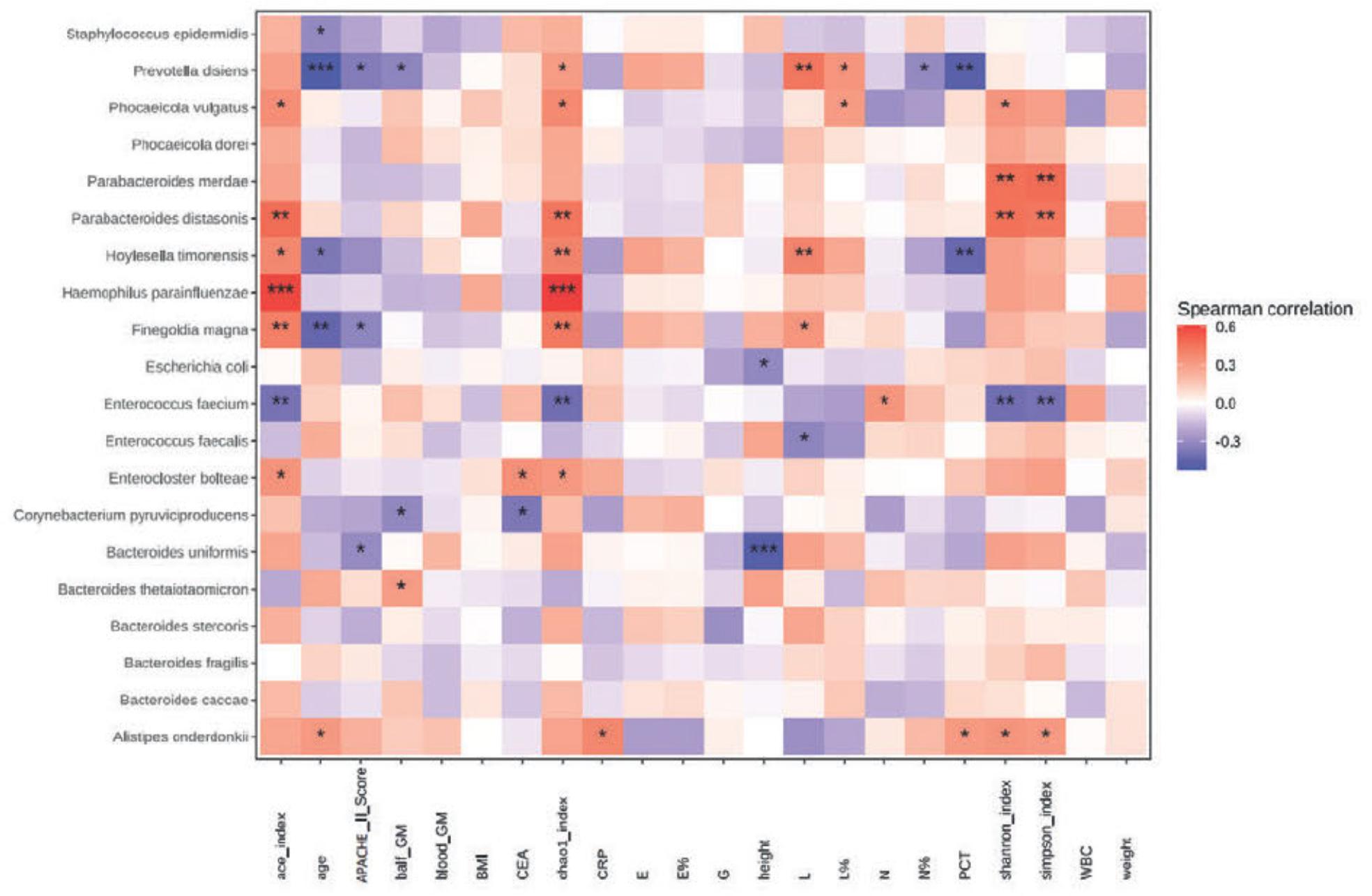
Spearman’s correlation analysis between clinical indicators and gut microbiota in patients with IPA.
The color depth in the heat map indicates the strength of the correlation: red indicates a positive correlation and blue indicates a negative correlation (* p < 0.05; ** p < 0.01; *** p < 0.001). APACHE II score - Acute Physiology and Chronic Health Evaluation II score; balf GM – bronchoalveolar lavage fluid galactomannan; blood GM – serum galactomannan; G – serum (1-3)-β-d-glucan; BMI – body mass index; CEA – carcinoembiyonic antigen; CRP – C-reactive protein; E – eosinophil count; E% – eosinophil percentage; L – lymphocyte count; L% – lymphocyte percentage; N – neutrophil count; N% – neutrophil percentage; PCT – procalcitonin; WBC – white blood cell, IPA – invasive pulmonary aspergillosis.
To assess the potential of gut microbiota as a biomarker for diagnosing IPA, we constructed a random forest model using 43 patients in the IPA group and 31 healthy individuals in the H group as the training set (Fig. 5A). Based on the mean decrease in the accuracy value of the random forest, the top five species were selected as markers to distinguish between patients with IPA and healthy individuals. These species included Clostridium fessum, Streptococcus pseudopneumoniae, Corynebacterium striatum, and two butyrate-producing bacteria, B. wexlerae and Faecalibacterium prausnitzii. As assessed via the receiver operating characteristic curve analysis, the model based on these five species achieved an area under the curve of 96.8%, indicating a good diagnostic performance (Fig. 5B).
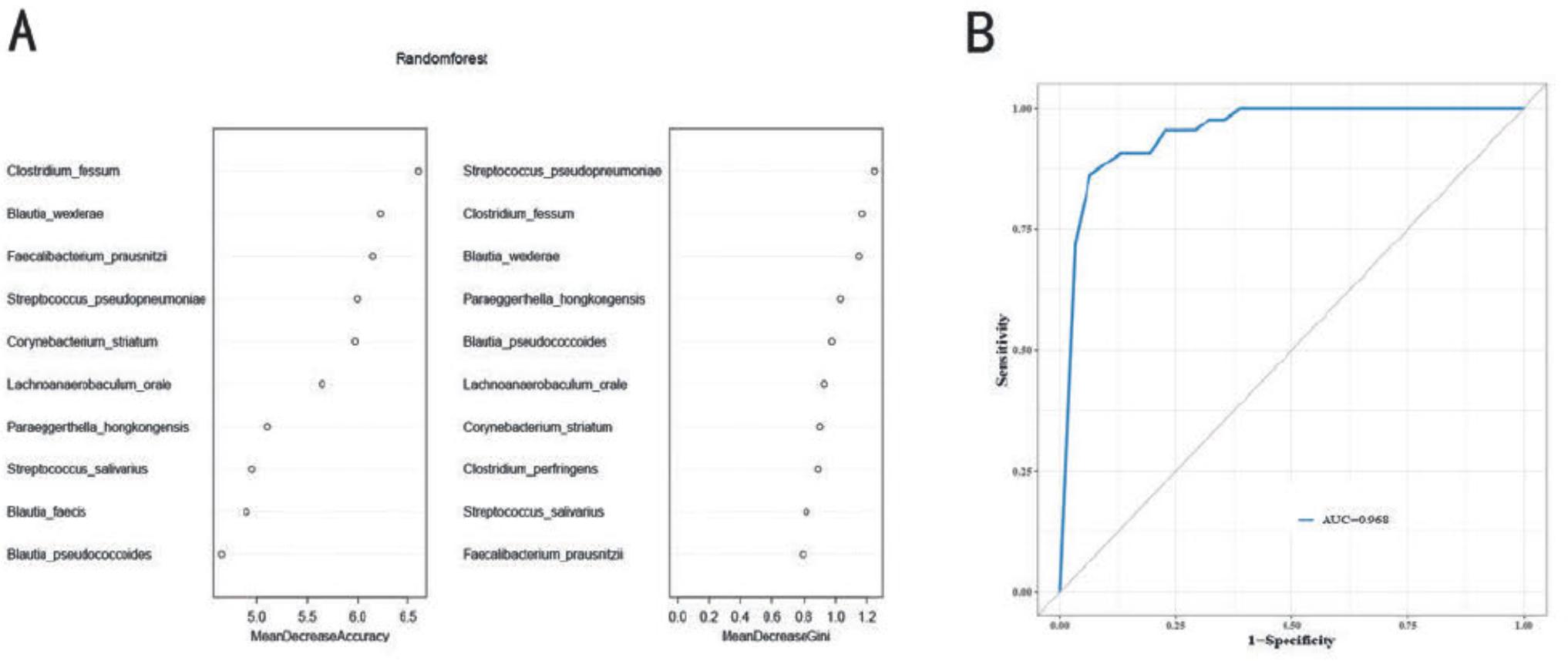
Random forest model used to distinguish patients with IPA from healthy controls.
A)Random forest mean decrease in accuracy and Gini; B) the area under the curve for the random forest model was obtained with the training set. IPA – invasive pulmonary aspergillosis.
Previous studies have found that the species diversity of gut microbiota in patients with community-acquired pneumonia or pulmonary tuberculosis is significantly lower than that in healthy individuals (Hu et al. 2019; Ren et al. 2020), and the reduction in alpha diversity is of particular value in predicting disease severity and prognosis (Hérivaux et al. 2022; Moutsoglou et al. 2023). In this study, compared to healthy controls, patients with IPA showed a significant decrease in the alpha diversity of gut microbiota and a significant difference in the beta diversity, indicating that the overall composition of the gut microbiota was different between the two groups and that pulmonary aspergillosis infection can cause disruption of the patient’s gut microbiota.
Our study found a significant increase in Ascomycota in IPA patients compared to the healthy control group. Previous studies have shown that Candida in Ascomycota can increase the concentration of prostaglandin E2 in the blood, promoting polarization of M2 macrophages and promoting the occurrence of allergic airway inflammation (Kim et al. 2014). Additionally, Ascomycota fungi have been implicated in immune activation and inflammatory responses during COVID-19 (Kusakabe et al. 2023). We hypothesize that increased Ascomycota in IPA patients may similarly promote systemic inflammation. However, the underlying mechanisms behind this potential inflammatory response remain to be further explored.
SCFAs are the products of dietary polysaccharides fermented by the gut microbiota, mainly including acetic acid, propionic acid, and butyric acid, which can regulate immune function and reduce the inflammatory response, thus reducing the incidence and severity of lung infections (Machado et al. 2021). Compared to the healthy control group, changes in gut microbiota in patients with IPA were characterized by a significant reduction in SCFA-producing species such as Faecalibacterium, Blautia, and Roseburia. In contrast, opportunistic pathogens, such as Enterococcus, Corynebacterium, Escherichia, Staphylococcus, Haemophilus, and Finegoldia, were significantly enriched. Among them, some species of Faecalibacterium, Roseburia, and Blautia participate in the production of SCFAs mainly to produce butyrate, which promotes the production of Tregs and exerts anti-inflammatory effects (Arpaia et al. 2013; Furusawa et al. 2013). Moreover, T cells may help the host prevent the occurrence of pulmonary aspergillosis (Hebart et al. 2002). Suppose metabolic analysis confirms that SCFAs in the gut of IPA patients are significantly reduced compared to healthy individuals. In that case, the reduced production of SCFAs caused by disturbances in gut microbiota may further affect the host immune and inflammatory responses to Aspergillus, thus promoting the progression of IPA. In addition, some strains of Faecalibacterium, Blautia, and Roseburia, recognized as potential next-generation probiotics (Sanders et al. 2019), may become new targets for IPA treatment.
One advantage of mNGS is that it can identify microorganisms at the species and strain level. Genetic and functional analyses on this basis can be used further to explore the role of specific species in diseases. Our results showed that the overall metabolic level of patients with IPA was significantly lower than that of healthy individuals. Additionally, we tentatively identified some relationships between species and metabolic pathways. B. wexlerae is usually significantly reduced in the guts of patients with intestinal inflammatory and metabolic diseases (Benítez-Páez et al. 2020; Hu et al. 2021; Park et al. 2023), and a similar phenomenon has been observed in patients with IPA. B. wexlerae has anti-inflammatory properties and can alter the host intestinal environment and lipid metabolism (Hosomi et al. 2022). Our study also showed that B. wexlerae was positively correlated with metabolic pathways, such as carbohydrate synthesis and degradation, CDP-diglycerol, and multiple amino acid biosynthesis. This is consistent with the results of previous studies by Hosomi et al. (2022). The decrease in the abundance of B. wexlerae in IPA patients is related to the downregulation of carbohydrate synthesis and degradation pathways, which may further lead to a reduction in the production of SCFAs, thereby exacerbating the inflammatory response. B. wexlerae also demonstrates a good ability to distinguish between patients with IPA and healthy individuals and exhibits a good diagnostic performance when combined with C. fessum, S. pseudopneumoniae, C. striatum, and F. prausnitzii.
We found that, in patients with IPA, Phocaeicola and Bacteroides, B. uniformis, Phocaeicola plebeius, and P. coprocola were significantly reduced. Notably, the decrease in B. uniformis was associated with an increase in the APACHE II score, which helps predict mortality rates in intensive care unit patients and sepsis patients (Sungono et al. 2022; Yang et al. 2024), suggesting that B. uniformis may be related to the severity of IPA. We also found that P. coprocola is positively correlated with metabolic pathways related to CDP-diglycerol biosynthesis. CDP-diglycerol is important intermediates in the synthesis of cardiolipin, and a decrease in cardiolipin synthesis may lead to mitochondrial dysfunction, resulting in the accumulation of oxygen free radicals and promoting inflammatory reactions in the body (Deng et al. 2009). Many Phocaeicola species were previously classified under the genus Bacteroides (García-López et al. 2019). Bacteroides play an important role in regulating the host immune system, including stimulating the anti-inflammatory factor interleukin (IL)-10 and suppressing the pro-inflammatory factor IL-17. This regulation helps maintain a balance between IL-10 and IL-17, thus exerting anti-inflammatory effects (Mazmanian et al. 2008; Zafar and Saier 2021). During Aspergillus infection, IL-10 is protective by inhibiting the inflammatory response (Grünig et al. 1997; Seo et al. 2005), while IL-17 promotes inflammation through neutrophil recruitment (He et al. 2022). Their role in balancing IL-10 and IL-17, Bacteroides, could be significant in the pulmonary anti-Aspergillus immune response. However, as this study is a retrospective study, we did not measure the concentrations of IL-10 and IL-17 in the two populations. Therefore, if it is confirmed that there are corresponding changes in these two inflammatory factors in IPA patients, it could further indicate that the reduction of Phocaeicola and Bacteroides weakens the overall anti-inflammatory ability of the gut microbiota in these patients.
E. faecium is a symbiotic bacterium found in the human gastrointestinal tract and is a common pathogen associated with hospital infections (Weiner et al. 2016). Cytolysins secreted by this organism can damage the human intestinal cell membrane and lead to infections (Gao et al. 2018). Previous studies have found that E. faecium significantly increased in the gut of severe COVID-19 patients (Xu et al. 2022), and this species was positively correlated with the number of neutrophils, which was accompanied by increased expression of genes related to platelet aggregation, endogenous coagulation, and neutrophil degranulation, indicating that E. faecium may affect the body’s innate immune response and coagulation function (Zhang et al. 2023). We observed a significant increase in E. faecium in the gut of IPA patients, which was positively correlated with neutrophil count. These findings, supported by previous research, suggest that the overgrowth of E. faecium promotes the host’s inflammatory response. Interestingly, an increase in E. faecium was also associated with a decrease in many metabolic pathways related to biosynthesis in patients with IPA, which needs further exploration in subsequent experiments.
In this study, we found that P. disiens was enriched in the intestines of patients with IPA, accompanied by an increase in the percentage of lymphocytes in the blood. Interestingly, this enrichment showed negative correlations with infection indicators such as blood PCT concentration and neutrophil percentage. These findings suggest that P. disiens may play a role in regulating immune and inflammatory responses. Previous research has shown that, in chronic inflammatory conditions such as periodontitis, bacterial vaginosis, and rheumatoid arthritis, Prevotella enrichment is associated with increased local or systemic inflammation (Larsen 2017); however, the organism can also produce SCFAs that play a beneficial role in human health (Chen et al. 2020). In patients with human immunodeficiency virus infection, the increase in Prevotella is accompanied by the activation of intestinal dendritic cells (DCs) and T cells, which regulate local immune responses (Dillon et al. 2014). In vitro experiments have shown that Prevotella can attenuate the level of Haemophillus influenzae-induced pro-inflammatory cytokine IL-12p70 in DCs (Larsen et al. 2012). It has also been observed to reduce NF-κB-driven inflammatory response in bronchial epithelial cells of patients with cystic fibrosis by inhibiting Pseudomonas aeruginosa’s growth (Bertelsen et al. 2021). However, the role of Prevotella in disease contexts remains debated (Iljazovic et al. 2021), and further experiments are necessary to validate our findings concerning Prevotella in our study.
This study has some limitations. First, our analysis was confined to patients from our research center, resulting in a small sample size. We developed a random forest model; however, no internal or external verification was performed. Thus, the accuracy and applicability of the proposed model need to be evaluated. Second, this is the first clinical study to comprehensively analyze gut microbiota characteristics in IPA patients, focusing on describing and analyzing observed clinical phenomena. While the potential roles of specific microbial species in IPA patients were inferred from prior research, no mechanistic experiments were designed to explore their functional mechanisms. This is a limitation of this article and a work that needs to be completed in the future. Finally, although we controlled for the effects of drugs and probiotics on the gut microbiota, we did not control for the diet of the two groups. Different dietary patterns impact the composition of gut microbiota in humans. For instance, adhering to a Mediterranean diet enhances the growth of probiotics such as Bifidobacteria and Lactobacilli in the gut. At the same time, vegetarians are associated with higher levels of Bifidobacteria and Prevotella. Conversely, diets high in fat and high sugar often lead to reduced diversity of gut microbiota, an increase in harmful bacterial species, and a decrease in beneficial bacterial species (Aziz et al. 2024). Therefore, the lack of control over dietary variables could impact the outcomes of this study.
We found an ecological imbalance in the gut microbiota of patients with IPA, primarily characterized by a reduction in species diversity. Changes in species abundance were characterized by decreased beneficial bacteria that produce SCFAs or exert anti-inflammatory effects, whereas some conditional pathogenic bacteria were enriched. In addition, we identified correlations between the gut microbiota, clinical indicators, and metabolic pathways, providing a direction for future research into these species’ exact mechanisms of action in patients with IPA. Moreover, a random forest model was used to identify biomarkers that could distinguish patients with IPA from healthy individuals. In summary, our findings provide new directions for diagnosing and treating patients with IPA.