
In 2015, the reclassification of class Tremellomycetes, which was established according to the molecularrather than morphology- and physiology-based identification, led to the separation of the Apiotrichum genus from the Trichosporon genus (Liu et al. 2015a). Apiotrichum cacaoliposimilis, previously known as Trichosporon cacaoliposimilis, was first isolated in Japan, likely from a soil sample collected from a vegetable farm. While the exact ecological niche of A. cacaoliposimilis remains debatable to date (Gujjari et al. 2011), it reportedly thrives on fermentation media and produces cellular lipids similar to those in natural cocoa butter (Yang et al. 2024). A. cacaoliposimilis was initially considered an oleaginous yeast-like fungus, which is capable of synthesizing and accumulating lipids up to 70% of its dry weight and is, therefore explored for valuable commercial products such as biofuels and cocoa butter equivalent (CBE) (Ghazani and Marangoni 2022).
The pathogenic potential of A. cacaoliposimilis is, however, not reported extensively in the clinical literature, which contains just one report on the isolation of A. cacaoliposimilis from a nail sample from a patient with onychomycosis (Noguchi et al. 2020). The knowledge regarding the diagnosis and treatment of A. cacaoliposimilis is limited to this single case report, and the complete genome of A. cacaoliposimilis is currently unavailable. Trichosporon is a genus related closely to A. cacaoliposimilis, and Trichosporon spp. have, to the best of the author’s knowledge, a pivotal role in different clinical infections. Trichosporon spp. resides occasionally on the skin surface and within the gastrointestinal environment in humans and comprises a diverse range of opportunistic pathogens that cause infections (Colombo et al. 2011; Noguchi et al. 2020) observed frequently in patients admitted to the intensive care units, particularly in patients with hematological malignancies or neutropenia and those undergoing broad-spectrum antibiotic therapy or invasive surgery (Francisco et al. 2019). The precise identification of the etiological agents at the taxonomic level is crucial, considering the variable antifungal drug susceptibilities of different Trichosporon spp. (Guo et al. 2019). The commercially available systems, such as API® 20 C AUX and Vitek® 2 Compact YST manufactured by bioMérieux (France), often yield uncertain results when rare species are involved (Guo et al. 2011). Moreover, the mass spectrometry-based libraries are enriched more with the commonly identified clinical pathogens, and their utility in detecting rare fungi is limited (Francisco et al. 2023). The internal transcribed spacer (ITS) sequencing facilitates a precise identification of yeast species (Langsiri et al. 2023), although it has not been adopted widely in clinical settings.
The existing literature on A. cacaoliposimilis is, to the best of the author’s knowledge, limited to specieslevel classification based on ITS and D1/D2 sequencing, biochemical responses, and drug susceptibility tests (Gujjari et al. 2011; Noguchi et al. 2020). The NCBI database contains 27 species within the Apiotrichum genus, including A. cacaoliposimilis (NCBI:txid105983). Among these species, at least six are reportedly associated with opportunistic infections (Taverna et al. 2014; Lara et al. 2019; Peng et al. 2019; Premamalini et al. 2019). However, previous studies lack a genomic analysis of A. cacaoliposimilis. Therefore, the present study was conducted to provide a reference for identifying the clinically rare pathogenic fungus A. cacaoliposimilis. Since the present report is a pioneer genome-wide report on this fungus, the meaningful annotation of the virulence genes of this fungus published in this report will serve as a basis for future research on the pathogenic mechanisms of this fungus. Further, the clinical protocols for treating this fungal infection will be elaborated based on the drug sensitivity test (DST).
A male patient aged 70 years presented to the emergency department of our hospital with the complaint of left lumbar discomfort and pyrexia (38°C). The patient also stated experiencing right upper abdominal pain, difficulty in urination, weak urine stream, and an increase in urinary frequency, urgency, and dysuria. A urological ultrasound revealed stones in the upper left ureter and left hydronephrosis. Physical examination elicited positive percussion pain above the left kidney. The patient was, therefore, provided with a primary diagnosis of left ureteral calculus accompanied by hydronephrosis and infection and a secondary diagnosis of UTI. Subsequently, the patient was admitted to the outpatient clinic of the hospital, where the laboratory tests revealed elevated white blood cell count (WBC) (17.58 × 109/l), high neutrophil percentage (95.4%), and increased levels of C-reactive protein (CRP) (173.85 mg/l), procalcitonin (PCT) (2.66 ng/ml), and creatinine (CR) (118.9 μmol/l). The other clinical findings were as follows: urinary leukocyte esterase, 1+/70 cells/μl; urinary erythrocytes, 289/μl; urinary leukocytes, 149/μl. In the period of hospitalization, the patient exhibited persisting pyrexia despite receiving the standard antibiotic therapy. Therefore, the mid-stream urine specimen was promptly sent to the clinical microbiology laboratory for inoculation and culture. After 48 hours of culture, no bacterial colony was detected, although fungal growth was noted. However, the mass spectrometry of the typical colonies with low confidence, smear microscopy, and biochemical analyses indicated the presence of an uncommon fungus, which was labeled UB0827 and analyzed further for identification. The subsequent susceptibility tests indicated sensitivity to antifungal medications. After that, the patient was administered voriconazole at an initial dose of 6 mg/kg every 12 h for a day, followed by a maintenance dose of the drug at 3 mg/kg every 12 h for three days. The treatment normalized the patient’s temperature on the third day of the antifungal treatment, after which the patient was discharged from the hospital.
Sabouraud agar with ATB® FUNGUS 3 (bioMérieux, France) was used in fungal drug sensitivity testing. Vitek® 2 Compact (bioMérieux, France), equipped with a YST identification card, was used for fungal identification. A MALDI-TOF mass spectrometer (Bruker Daltonics GmbH & Co., Germany) was employed to validate the identification. Gram staining, lactophenol cotton blue staining, fluorescence staining, and hexamine silver staining were performed using the respective reagents from Zhuhai Baso Biotechnology Company (China).
Microbial culture was performed using the Sabouraud agar medium at 35°C for 72 h. The isolates were then analyzed using Gram staining, lactophenol cotton blue staining, fluorescent staining, and hexamethylamine silver staining. The stained samples were photographed under a microscope for further analysis.
Three to five colonies developed on the Sabouraud agar medium culture were isolated and resuspended in a 0.45% solution of sterile saline, followed by adjusting the suspension culture density to the McFarland concentration of 1.8–2.2. The suspension culture was then transferred to a tube containing the YST identification card, and incubation was performed in the Vitek® 2 Compact system for 24 h. A total of three independent replicates were used for the test.
The fungal samples were placed on the designated regions on a VITEK® MS-DS target slide and spread uniformly using a 1 μl inoculation loop according to the manufacturer’s instructions. Subsequently, 0.5 μl of the VITEK® MS-FA reagent was placed on each designated spot, and after 1–3 min, 1 μl of the VITEK® MS-CHCA matrix solution was added. After drying, the samples were processed using the VITEK® MS instrument.
ATB® FUNGUS 3 was utilized to select colonies from the Sabouraud agar medium, following the manufacturer’s instructions. The selected colonies were then suspended in 0.85% NaCl solution, and the McFarland turbidity was adjusted to 2.0 using the McFarland turbidimeter. Next, 20 μl of the resulting culture solution was transferred to ATB® FUNGUS 3. After adding the solution to the sealed wells containing the antifungal drug, it was incubated at 35.8°C for 48 h. Fungal growth in each well was compared to that in the blank control well to determine the minimum inhibitory concentration.
The fungal lysates for ITS sequencing were obtained using the TaKaRa lysis buffer for the microorganism provided in the PCR kit (Code No. 9164; Takara Bio Inc., Japan) according to the manufacturer’s instructions. First, a single colony was lysed in a microtube by adding 50 μl of the lysis buffer. Next, the resulting lysate was vortexed thoroughly followed by denaturation at 80°C for 15 min and centrifuged at 1,000 × g to precipitate cell debris. The resulting clear supernatant was subjected to a polymerase chain reaction (PCR) to amplify the DNA content for further analysis. The PCR reaction mixture contained 1–5 μl of the lysate supernatant, 25 μl of Premix Taq, 1 μl of the Forward Primer (10 pmol/μl), and 1 μl of the Reverse Primer (10 pmol/μl), and was supplemented with sterile nuclease-free water to reach a total volume of 50 μl. The primers used in the PCR were ITS1 (5’-TCCGTAG-GTGAACCTGCGG-3’) and ITS4 (5’-TCCTCCGCT-TATTGATATGC-3’), which were designed by Gujjari et al. (2011). The PCR products were confirmed using 1% agarose gel electrophoresis, and the target sequences were recovered using the TIANGEN gel recovery kit (TIANGEN Biotech(Beijing)Co., Ltd., China). The recovered sequences were analyzed through Sanger sequencing (Beijing Prime Biotechnology Co., China).
Genomic DNA was extracted from a frozen colony using the cetyltrimethylammonium bromide protocol (Kim et al. 2010). The DNA samples were then fragmented randomly using a Covaris ultrasonic crusher (Covaris, LLC., USA). Library preparation involved end repair, addition of the A-tail and sequencing junction, PCR, fragment screening, and purification. Multiple libraries with the same effective concentration and the desired downstream data volume were pooled and subjected to double-end sequencing (Illumina, Inc., USA). The quality control of the sequencing data involved the removal of the reads with > 40% low-quality bases (quality ≤ 20), ≥ 10% unknown bases, and 15 bp overlap with adapter sequences. The sequencing data were then assembled using SOAP denovo (v2.04) (Luo et al. 2012) with GapCloser (v1.12). Sequences < 500 bp in length were filtered out, and the remaining sequences were subjected to a statistical analysis and gene prediction. Genomic completeness was assessed based on the BUSCO score (Simão et al. 2015), and the ratio of the Illumina reads was mapped back to the genome.
The ITS and whole genome sequences of various species (Table SI) were downloaded from the NCBI genome database. The ITS sequences obtained from Sanger sequencing were aligned with those downloaded from the NCBI genome database using Muscle5 (Edgar 2022). Phylogenetic trees were then constructed using the maximum likelihood method using IQ-tree (v2.0) (Minh et al. 2020).
Mugsy (v1.2.3) (Angiuoli and Salzberg 2011) was employed to identify and extract the homologous regions between the assembled genome and reference genomes from the selected species, which were annotated in the MAF format. These files were subsequently converted to the FASTA format and aligned using MAF to FASTA (v1.0.1) in Galaxy (Galaxy Community 2022). The homologous regions with high-quality alignments were extracted using trimAl (v1.4.1) with default parameters (Capella-Gutiérrez et al. 2009), and the gaps were removed using Seqkit (v2.6.1) (Shen et al. 2016; 2024). The resulting sequences were compared with default parameters using MAFFT (v7.520) (Rozewicki et al. 2019). Finally, a maximum likelihood tree was constructed using RAxML (v8.2.13) (Stamatakis 2014). The bootstrap confidence intervals were calculated based on 1000 replicates.
RepeatMasker software (v open-4.0.5; http://www.repeatmasker.org) was employed to identify and mask the scattered repeat sequences. Tandem repeats in the DNA sequences were searched using Tandem Repeats Finder (v4.07b) (Benson 1999). The transfer RNAs and ribosomal RNAs were predicted using tRNAscan-SE (v1.3.1) (Lowe and Eddy 1997) and RNAmmer (v1.2) (Lagesen et al. 2007), respectively. Comparative annotation was performed using Rfam (Kalvari et al. 2021) to identify the bacterial small RNAs, small nuclear RNAs, and micro RNAs. CMsearch (v1.1rc4) was used with default parameters to confirm the bacterial small RNA annotations.
The coding genes in the newly sequenced genome were predicted using Funannotate (v1.8.1) (Palmer and Stajich 2020). The protein sequences encoded by these predicted genes were compared against those in the functional databases, such as GO, KEGG, NR, and CAZy using Diamond (v2.0.11.149) (Buchfink et al. 2021) and an e-value threshold of ≤ 1e-5. The sequences with ≥40% identity and ≥40% coverage were then selected and annotated. The signal peptides and transmembrane structures were identified using SignalP (v4.1) (Petersen et al. 2011) and TMHMM (v2.0c) (Sonnhammer et al. 1998; Krogh et al. 2001), respectively, which facilitated the prediction of the secreted proteins. AntiSMASH (v2.0.2) (Blin et al. 2013) was utilized to predict the secondary metabolic gene clusters in the genome.
The genome sequences of Apiotrichum, Trichosporon, Cutaneotrichosporon, and Vanrija were obtained from the NCBI genome database. The downloaded dataset included the reference sequences of 81 species (Table SI). Since gene and protein annotations were lacking for most of these species, gene prediction was performed using Funannotate (v1.8.1). Gene clustering analysis was then performed using OrthoMCL and OrthoVenn3 (https://orthovenn3.bioinfotoolkits.net) with default parameters. Synteny analysis was also conducted using MCScanX in TBtools (Chen et al. 2023). Inter-species comparisons were conducted by selecting the representative species from closely related genera, including Apiotrichum (A. porosum DSM 27194, A. brassicae JCM 1599, A. mycotoxinovorans GMU1709, and A. gracile JCM 10018), Trichosporon, and Cutaneotrichosporon (T. asahii CBS 2479 and C. oleaginosum IBC 0246). Intra-node comparisons were conducted by focusing on Apiotrichum akiyoshidainum HP2023 and Apiotrichum laibachii JCM 2947. The primary objective of this comparative genomic analysis was to elucidate the intricate evolutionary relationships among these genomes.
Fungal virulence and resistance genes were identified, and the pathogenicity-related intraspecies and intergeneric genetic differences were determined using BLASTp in Diamond (v2.0.11.149) with the following parameters: e-value 1e-5; query coverage 40; identity 40; ultra-sensitive. Protein sequences were mapped to several databases, including the Mycology Antifungal Resistance Database (Nash et al. 2018), Pathogen-Host Interactions database (PHI-base) (Urban et al. 2022), Comprehensive Antibiotic Resistance Database (Alcock et al. 2023), and the database of fungal virulence factors. Wilcox test was performed using SPSS® 28.0 software (IBM® SPSS® Statistics for Windows, version 28.0, IBM Corp., USA) to determine the inter-group differences. The results were visualized using TBtools.
On the third day of incubation at 35°C for strain UB0827, the largest creamy white colonies, each 35 mm in diameter, with a fuzzy texture and irregular edges, were observed. The surface folds of these colonies had a ridged center on Sabouraud’s medium (Fig. 1A). After Gram staining, dark purple subglobular, ovoid, oval cells approximately 3–6 μm in diameter and the formation of potential arthrospores from violet mycelium shedding was noted at 1000 × magnification (Fig. 1B). Lactophenol Cotton Blue staining highlighted the blue-stained oval, short rod-like structures and translucent structures with granular-like organelles inside spores (Fig. 1C). The hexamethylene-silver staining revealed mycelium in a paler greenish color, highlighting the junctions where the distinct potential arthrospores were connected (Fig. 1D). When observed under a fluorescence microscope, the fungal hyphae and budding spores exhibited a distinctive feature of emitting a vibrant blue fluorescence under UV light (Fig. 1E).
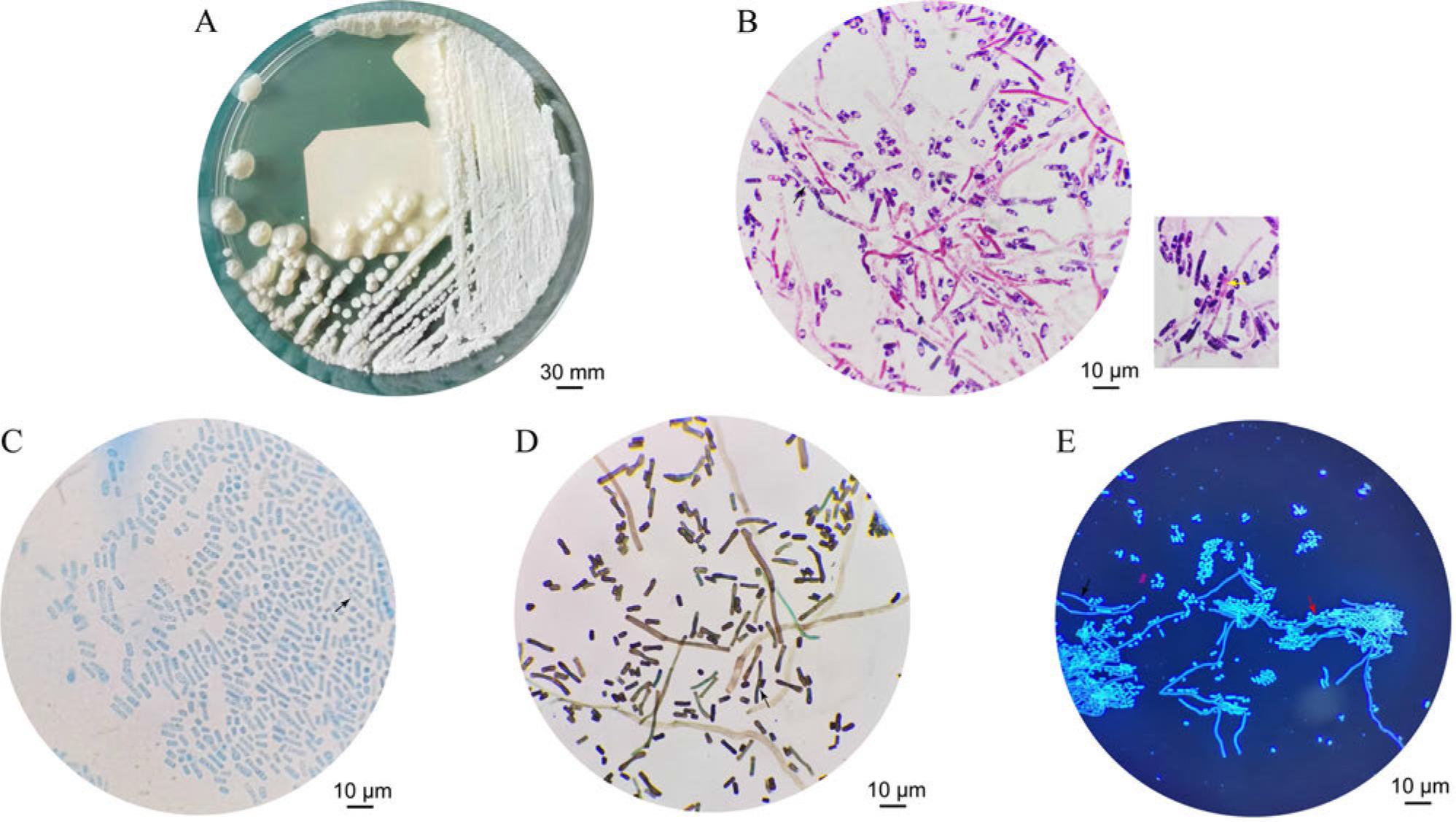
Morphology of the UB0827 strain: A) image after incubation on Sabouraud’s medium for 3 days; B) Gram staining under the microscope (1000× magnification); C) lactophenol cotton blue staining under the microscope (1000× magnification); D) hexamethylamine silver staining under the microscope (1000× magnification); E) fluorescent staining under the microscope (1000 magnification). Black arrows indicate the fungal filaments. Yellow arrows indicate the potential arthrospores. Red arrows indicate the budding spores.
The results of the biochemical tests conducted using Vitek® 2 Compact are presented in Table I. The identified potential suspects were Cryptococcus laurentii (Papiliotrema laurentii) and T. asahii. A comparison of the biochemical characteristics of A. mycotoxinivorans GMU 1709 and A. cacaoliposimilis ATCC® 20505™ (Gujjari et al. 2011; Peng et al. 2019) revealed that strain UB0827 exhibited weak responses in saccharification, nitrate, and citrate assimilation reactions. Moreover, the mass spectrometry analyses conducted using the MALDI-TOF technology did not support biochemical identification, and the confidence level was relatively low.
The following are the different/important biochemical reactions between strains Apiotrichum cacaoliposimilis UB0827, ATCC® 20505™ and Apiotrichum mycotoxinovorans GMU1709.
| Biochemical reaction | Strain | ||
|---|---|---|---|
| UB0827 | ATCC® 20505™ | GMU1709 | |
| L-lysine-arylamidase | − | + | − |
| Erythritol assimilation | − | − | − |
| Glycerol assimilation | − | + | + |
| β-N-Acetyl-glucosaminidase | − | + | + |
| Arbutin assimilation | − | + | − |
| Methyl-α-D-glucopyranoside assimilation | − | + | − |
| D-Raffinose assimilation | − | − | + |
| PNP-N-acetyl-β-D-galactosaminidase 1 | − | − | + |
| D-Melibiose assimilation | − | D | + |
| D-Melezitose assimilation | − | + | − |
| L-Sorbose assimilation | − | + | − |
| Xylitol assimilation | − | + | − |
| D-Sorbitol assimilation | − | / | + |
| Urease | + | + | + |
| D-Trehalose assimilation | + | + | + |
| Nitrate assimilation | − | + | + |
| D-Galacturonate assimilation | + | − | + |
| Esculin hydrolysis | − | / | + |
| DL-Lactate assimilation | + | − | + |
| Citrate (sodium) assimilation | − | D/W | + |
Assimilation reactions: D-galactose (+), D-glucose (+), lactose (+), D-cellobiose (+), D-maltose (+), saccharose/sucrose (+), L-arabinose (+), D-xylose (+), 2-keto-D-gluconate (+), D-gluconate (+)
+ – positive, - – negative, D – delayed positive, W – weak positive
A comparison of the sequenced ITS regions using BLAST yielded several results with > 99% identity and ≤ 3 gaps. The neighbor-joining tree (Fig. S1a) for the top 10 results, constructed using the Distance tree function of Web results, revealed a close relationship between the species isolated in the present study and A. cacaoliposimilis. The maximum likelihood tree (Fig. S1b) constructed based on the ITS sequences revealed that the isolated strain UB0827 was closely related to A. cacaoliposimilis and A. akiyoshidainum. Even after 5,000 repetitions of random sampling, the best Bootstrap value could not be obtained because UB0827 was situated at the wrong branch relative to A. akiyoshidainum.
The antifungal susceptibility testing conducted using ATB® FUNGUS 3 revealed the following results in terms of the minimum inhibitory concentrations of multiple antifungal drugs: amphotericin B, 0.5 μg/ml; 5-fluorocytosine, 2 μg/ml; triazole antibiotic fluconazole, 2 μg/ml; itraconazole, 0.12 μg/ml; voriconazole, 0.06 μg/ml; posaconazole, 6 μg/ml; echinocandin antibiotic, 8 μg/ml (Table II).
Antifungal susceptibility of Apiotrichum cacaoliposimilis UB0827.
| Antifungal susceptibility (MICs, μg/ml) | ||||||||
|---|---|---|---|---|---|---|---|---|
| 5FC | AMB | FLU | POS | VOR | ITZ | ANID | MICA | CASP |
| 2 | 0.5 | 2 | 0.06 | 0.06 | 0.12 | 8 | 8 | 8 |
5FC – 5-fluorocytosine, AMB – amphotericin B, FLU – fluconazole, POS – posaconazole, VOR – voriconazole, ITZ – itraconazole, ANID – anidulafungin, MICA – micafungin, CASP – caspofungin, MICs – Minimum Inhibitory Concentrations
Genomic data of approximately 2.56 Gb in quantity were generated through Illumina sequencing. The 15-mer analysis was then conducted to estimate the actual and fitted genome sizes, which were 25.6 Mb and 26.27 Mb, respectively, using a k-mer depth of 33.31 × (average sequence depth 69×). The scaffold N50 was approximately 406.6 Kb. The average GC content was approximately 60.83%, and 88.2% of the complete BUSCO genes in the genome of A. cacaoliposimilis UB0827 belonged to the expected class Tremellomycetes (Fig. S2).
Using RepeatMasker and Tandem Repeats Finder, a total of 2,525,374 bp of repetitive sequences were detected in the evaluated genome, including 681,841 bp of long terminal repeats, 251,865 bp of long scattered repeats, 253,304 bp of short, scattered repeats, 271,919 bp of DNA repeats, 124,837 bp of minisatellite DNA, and 25,926 bp of microsatellite DNA. Homologybased and ab initio prediction analyses revealed 7,161 protein-coding genes in the evaluated genome, with an average length of approximately 1,702.19 bp. Among these genes, 6,734 genes encoded messenger RNAs and 427 genes encoded transfer RNAs. The average number of exons in these genes was approximately 4.35, with an average length of 312.4 bp. The average length of the proteins encoded by these genes was approximately 467.34 amino acids. Eleven small nuclear RNAs could be aligned, although no validated ribosomal RNAs were predicted. Annotation with the Gene Ontology database revealed 4,549 gene classes, including 8,692 in the category of biological processes, 5,663 in the category of cellular components, and 5,422 in the category of molecular functions. The weight of each of these classes is presented in Fig. 2. Further, 3,270 genes were annotated in the Kyoto Encyclopedia of Genes and Genomes, and most of these genes were related to infectious diseases. In addition, these genes were involved in various pathways, most related to carbohydrate and amino acid metabolism (Fig. S3a).
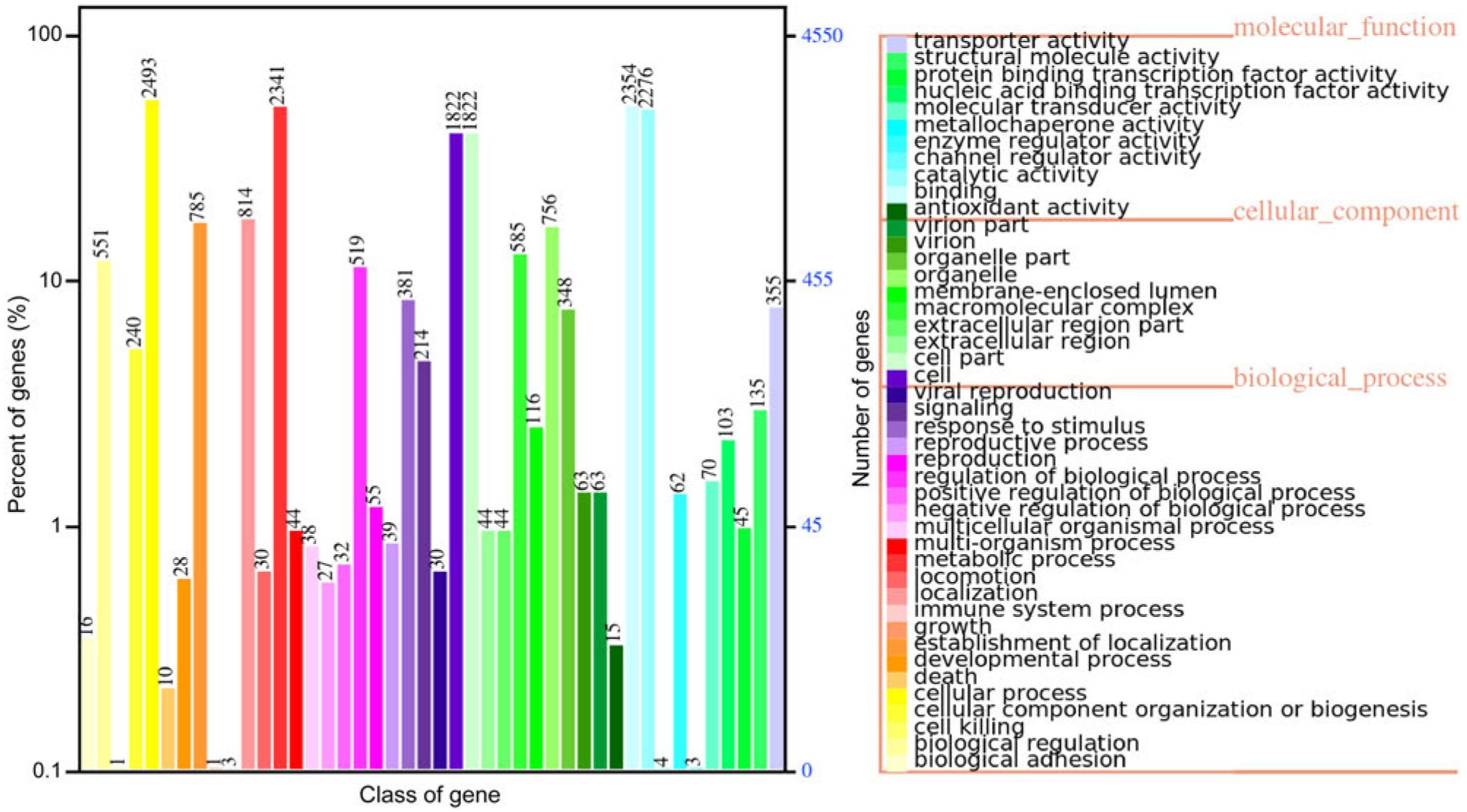
The legend on the right side of the figure lists the functional classifications of the Gene Ontology (GO) annotations for the corresponding samples under the three categories of GO ontology classifications. The left side of the figure depicts the distribution of a number of GO functional analyses on the annotations. The horizontal axis represents the number of genes, displayed in different colors corresponding to the different types mentioned in the legend on the right. The vertical axis presents the GO functional classification.
The genome-wide phylogenetic tree (Fig. 3) confirmed that the isolated A. cacaoliposimilis belonged to the Apiotrichum genus, consistent with the ITS sequence alignment results. The tree, with a Bootstrap value of 100, obtained from 1,000 random replicates, placed the isolated strain at the same node as that of A. akiyoshidainum HP2023 and A. laibachii JCM 2947. The branch lengths distinguished the isolated strain from A. akiyoshidainum HP2023, indicating that this fungus belonged to the A. cacaoliposimilis species. Moreover, the evolutionary relationship noted between A. akiyoshidainum HP2023 and T. cutaneum ACCC 20271 was consistent with the report of Peng et al. (2022). Accordingly, a highly reliable evolutionary relationship of the isolated fungus was established, named A. cacaoliposimilis UB0827. However, since this is the first strain to be sequenced without intra-specific whole-genome sequence comparisons, further confirmation of this phylogenetic relationship is necessary.
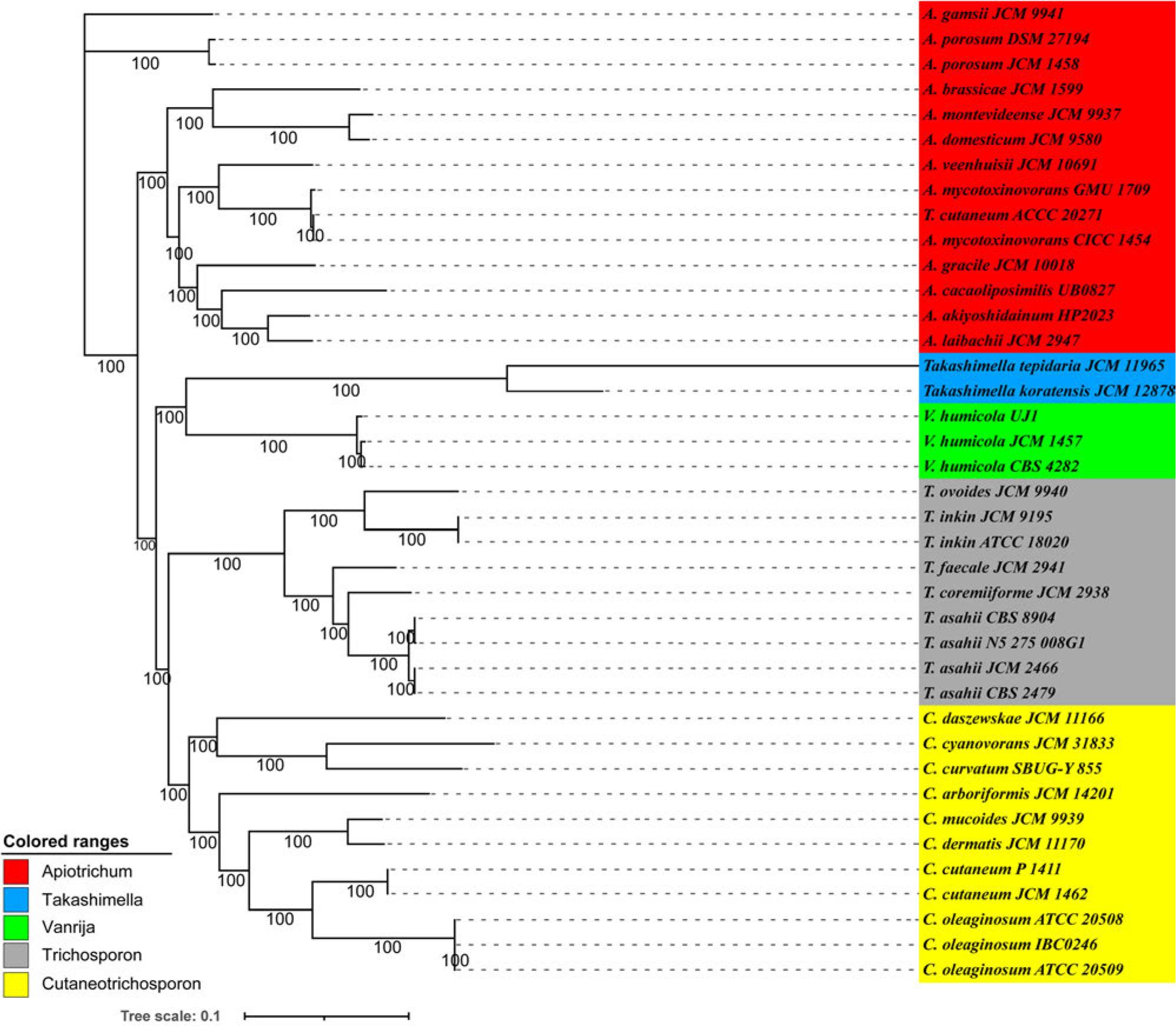
A phylogenomic analysis of the thirty-seven members of the Trichosporonaceae family, conducted using the maximum likelihood (ML) method. The ML tree was constructed based on the concatenated non-gapped sites from multiple whole-genome alignments. The branch length of this tree represented the evolutionary distance on the tree scale. The phylogenetic analysis was conducted using raxmlHPC-PTHREADS-AVX (v8.2.13), utilizing the GTRGAMMA model and 1,000 bootstrap replicates. The genera Apiotrichum, Trichosporon, Cutaneotrichosporon, and Vanrija within the Trichosporonaceae family and the genus Takashimella (outgroup) in Tetragoniomycetaceae are highlighted in red, gray, yellow, green, and blue, respectively.
Markov clustering analysis identified 454,970 protein sets from the 81 strains (Table SI) belonging to the four genera of Trichosporonaceae. Among these sets, 98.2% (446,773) of the protein sets were clustered into 15,425 orthologous groups, with 319 orthogroups specific to certain species. The evolutionary positions among 81 species in the genome-wide phylogenetic tree were validated by constructing the maximum likelihood tree based on the homologous gene families (Fig. S4). The Venn diagram (Fig. 4A) analysis and comparison of A. cacaoliposimilis UB0827 with the four genera of Trichosporonacea revealed that the A. cacaoliposimilis strain UB0827 shared the highest number of orthogroups with the Apiotrichum genus (Apiotrichum: 5,161 >Cutaneotrichosporon: 4,822 >Trichosporon: 4,007 >Vanrija: 3,784). This finding is supported by the non-redundant protein database annotations, according to which the homologous proteins of A. porosum are more numerous than any other species (Fig. S3b). Further, Fig. 4B presents the comparison of the four branches (A. cacaoliposimilis UB0827, A. akiyoshidainum HP2023, A. gracile JCM10018, and A. laibachii JCM2947) to the orthogroups, illustrating the intersections of 4,941, 4,529, and 5,302, respectively. These results supported the observed genome-wide phylogenetic relationships.
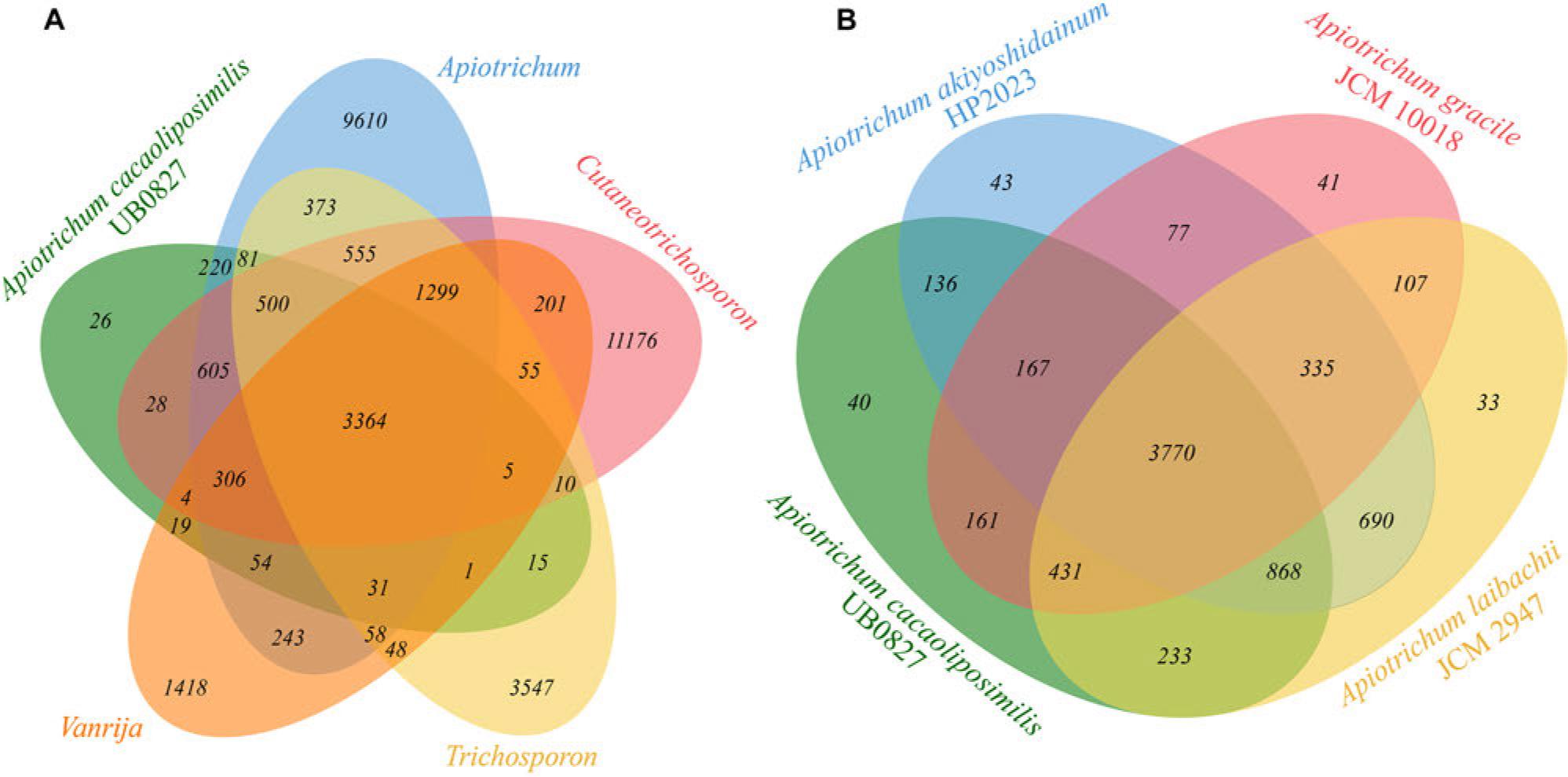
A) Venn diagram illustrating the relationships between A. cacaoliposimilis UB0827 and the different Trichosporonaceae genera based on the 6656 orthogroups; B) Venn diagram illustrating the relationships among different species positioned on the same node.
Genomic synteny block analysis discerned the evolutionary relationships among the different strains, revealing synteny among A. cacaoliposimilis UB0827, A. gracile JCM10018, and A. laibachii JCM2947 (Fig. 5). Notably, A. cacaoliposimilis UB0827 exhibited a considerable degree of synteny with A. mycotoxinivorans GMU 1709. However, this synteny was not as extensive as that demonstrated with A. gracile JCM10018, affirming their evolutionary linkage. In contrast, A. akiyoshidainum HP2023 exhibited lesser synteny. A separate comparison of A. cacaoliposimilis UB0827, A. gracile JCM10018, and A. laibachii JCM2947 revealed 2154, 1557, and 3683 synteny blocks, respectively (Fig. S5), demonstrating the evolutionary connections among these four. The observed differences could be attributed mainly to the quality of genome assembly. These findings corroborated the evolutionary affiliation of A. cacaoliposimilis UB0827 within the Trichosporonaceae family.

The synteny analysis performed at the genomic level for strain UB0827 and the representative strains of different nodes. The number displayed between the strains indicates the number of syntenic blocks, which become increasingly dense when moving from top to bottom. The straight line exists due to the poor assembly quality of the HP2023 strain.
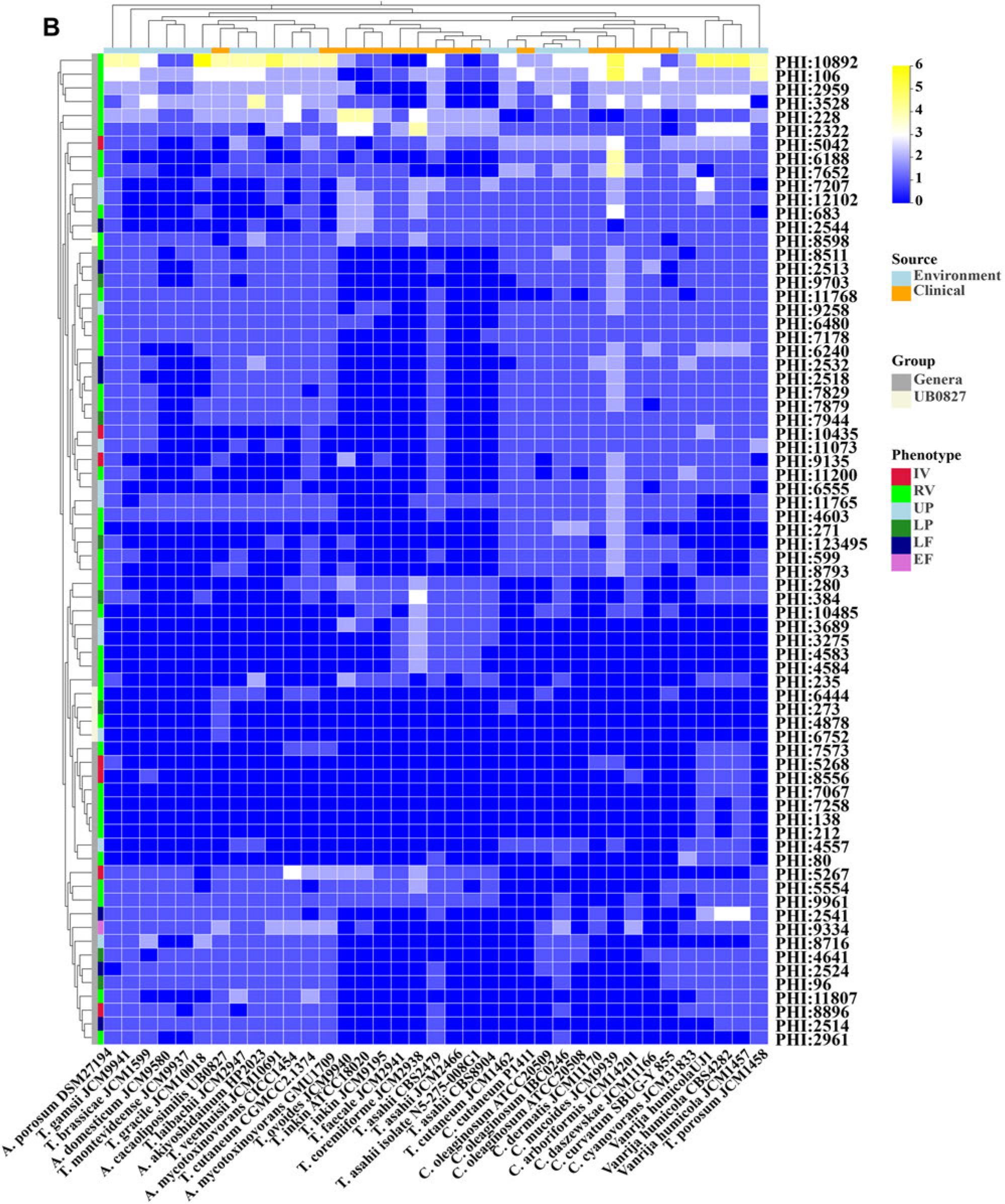
PHI-base is a gene-centric database of paramount significance, which curates the experimentally validated virulence, pathogenicity, and effector genes sourced from diverse pathogens. Each gene in this database has been assigned a distinctive PHI-base accession number, facilitating a streamlined identification and retrieval process. This comprehensive database has an expansive array of included pathogens, encompassing interactions with a broad spectrum of hosts, due to which it assumes a pivotal role in unearthing the pathogenicity-linked genes from emergent opportunistic pathogens (Urban et al. 2022). The identification of such genes would facilitate the identification of potential targets for interventions to combat the harmful influence of these pathogens. The current iteration of PHI-base version 4.16 boasts an impressive compendium documenting an astounding 9,666 genes and 21,676 interactions linked intrinsically to pathogenicity. A. cacaoliposimilis is an emerging opportunistic pathogen that exhibits pathogenicity through invasive manipulation and immunodeficiency (Noguchi et al. 2020). A comparison of the PHI databases under the ULTRA SENSITIVE mode of blast search conducted in the present study revealed 741 PHI homologous genes, which was higher than the average in all compared strains (704). Further analysis with the terms “Primates”, “Birds”, “Rabbits & hares”, “Nematodes”, “Flies”, “Bony fishes”, and “Rodents” used under the category of interacting hosts identified 296 PHI homologous genes (6% hypervirulence, 9% lethal, 14% loss of pathogenicity, 18% unaffected pathogenicity, and 51% reduced virulence). These organisms were explicitly selected as they have frequently been utilized in various studies as models to understand human diseases better.
A higher number of PHI genes were detected in the environmental isolate A. porosum DSM27194. Grouping of the strains based on their origin (environmental and clinical) and the statistical analysis based on the Wilcoxon rank-sum test revealed non-significant differences (p> 0.05). The inter-species comparison of A. cacaoliposimilis UB0827 was performed using two grouping methods: one based on A. cacaoliposimilis UB0827 and other species and the other based on genera. A comparison of the differences between these two groups identified 215 significant PHI-base accessions (210 intergeneric and five inter-specific differences). Specifically, 72 intergeneric (p < 0.001) and five interspecific (p < 0.05) differences in PHI were noted (Fig. 6B). The PHI accessions of the inter-specific differences included PHI: 273, PHI: 4878, PHI: 6444, PHI: 6752, and PHI: 8598, which encode the SAGA complex species spt3 protein (spt3), a putative serine/threonine protein phosphatase type 2A (ppg1), NADH-ubiquinone oxidoreductase 299 kDa subunit (Complex I NADH oxidoredutase), F-box Protein Fbx15 (grrA), and chitin deacetylase (cda1, except for UB0827), respectively. The inter-generic comparisons were conducted by compiling a detailed catalog (Table SII) of all PHI genes in UB0827, including the four inter-species differential genes. While 60% of the causative genes were revealed to be associated with lung tissue, 40% were associated with metabolic enzyme reactions. Among the highly pathogenic genes, PHI: 5267 and PHI: 8896 were specific to the Apiotrichum genus and encoded epimerase (ugeB) and phosphatidyl-N-methylethanolamine N-methyltransferase (pem2), respectively. PHI: 9135, which encodes serine/threonine-protein kinase (atg1), was more common in the Cutaneotrichosporon genus. Moreover, considering the syntenic relationship, 64.86% (192 of 296) of the pathogenic genes were common to the pathogenic strains UB0827 and GMU1709 (Fig. 6A) and included nine genes associated with high virulence and 19 lethal factors. In addition, nine hypervirulence genes (atg1, tufa, pkr1, aspB, CaPph3, aspC, nth1, mgt2, and GroEL) and six lethal factors (gfa1, met2, pmt1, pmt4, pri1, and glcA) were specific to UB0827.
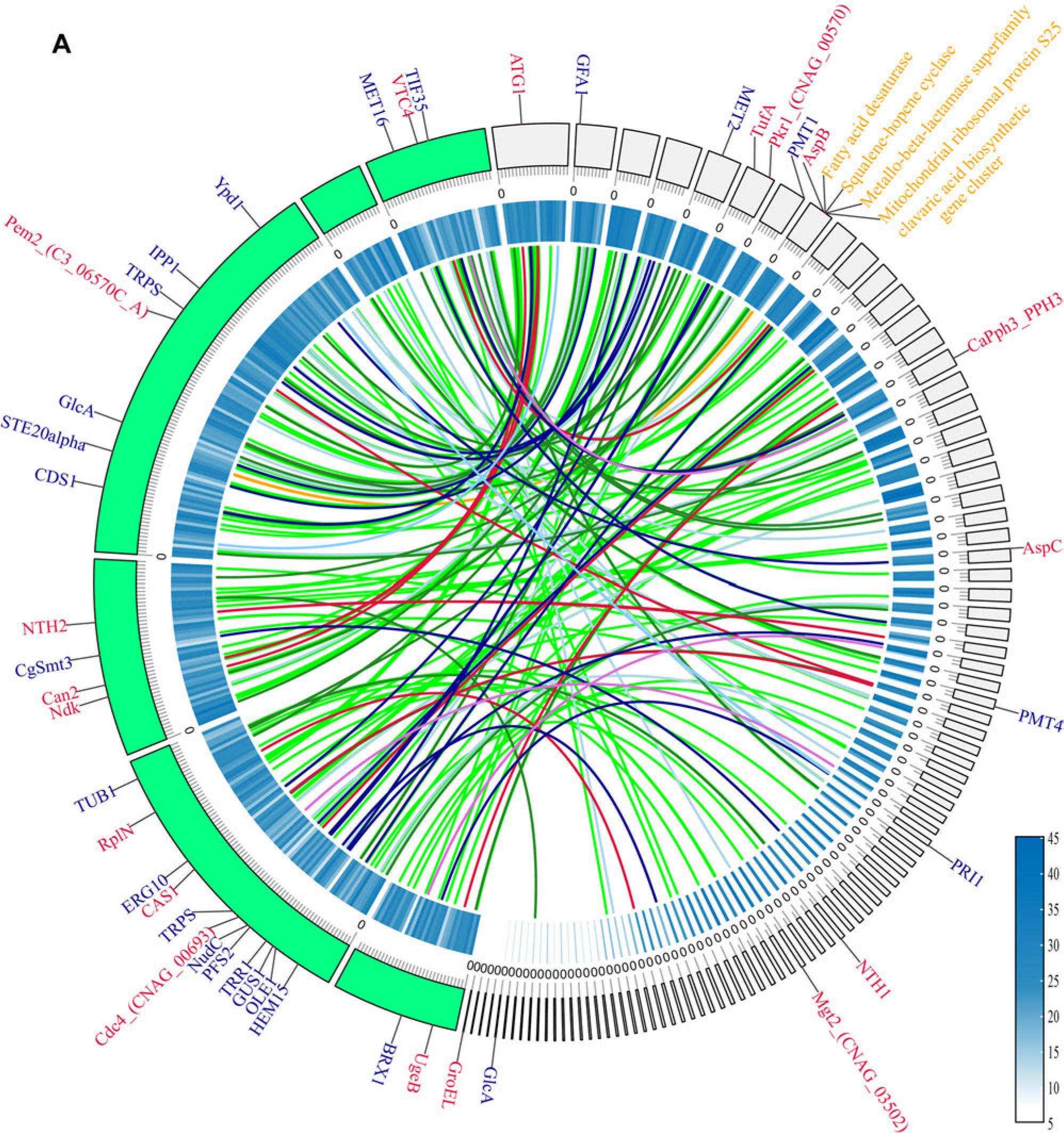
A) The outermost circle represents the chromosome skeleton, with strain GMU1709 depicted in green and strain UB0827 depicted in light grey. The inner circle indicates gene density, with a gradient from white to blue indicating an increase in the number of genes. Syntenic pairs indicate the shared pathogenic genes. The genes depicted on the left skeleton are shared genes, while the genes depicted on the right skeleton are the genes specific to A. cacaoliposimilis. The colors of the genes indicate their respective phenotypic classifications. B) A total of 77 disease-causing genes are depicted, with 72 of these genes differing between the various genera and 5 genes differing between species. The outermost color on the left corresponds to the grouping, while the innermost color represents the phenotypic classification of the disease-causing genes. The top hints at the origin of the isolated strain. The gradient from dark blue to yellow indicates an increase in the number of genes.
The first strain of A. cacaoliposimilis, which could produce lipids resembling natural cocoa butter, was isolated from the soil (Gujjari et al. 2011). The fungi of this species are considered essential producers of biological lipids (Silva et al. 2023) and cocoa butter alternatives (Yang et al. 2024). The predominant method for the clinical detection of A. cacaoliposimilis involves culturing for morphological identification, followed by biochemical analyses for species determination. However, a limitation of this approach is that it does not identify and differentiate the closely related species precisely. Considering the distinct pathogenic effects and drug resistance profiles of various fungal species within the Apiotrichum genus, accurate identification is crucial for the clinical prevention and treatment of infections caused by these fungi. Mass spectrometry and molecular identification have facilitated more access to precise species identification. Mass spectrometry is instrumental in identifying Trichosporon genus, although it exhibits lower effectiveness for Apiotrichum genus (de Almeida et al. 2017; Ahangarkani et al. 2021). Over the past decade, the focus of molecular identification analyses has shifted from phenotypic to molecular markers. For instance, basidiomycetous fungi may be identified based on the GC molar ratio of their nuclear DNA, which ranges from 50% to 70% (Kurtzman 2006). ITS sequences are invaluable for accurate species identification, as demonstrated previously in studies conducted with yeast microbiota (García-Béjar et al. 2023). In the present study, first-generation sequencing technology was utilized to retrieve the ITS segments of a fungus that closely resembled A. cacaoliposimilis ATCC® 20505™ (identities 485/488, gaps 3/488; Fig. S1a) and was different from A. akiyoshidainum M8 (formerly known as Trichosporon akiyoshidainum, identities 482/485, gaps 2/485). Since the nucleotide sequence differences between A. cacaoliposimilis and A. akiyoshidainum were revealed to be mostly present in the D1/D2 region (Gujjari et al. 2011), a phylogenetic tree was constructed using the ITS regions. The tree validated that the isolated fungus was closest to A. akiyoshidainum or A. cacaoliposimilis, although with a lower bootstrap value, possibly due to the shorter sequences and the influence of the conserved loci (Paradis et al. 2023). Differential evolution or horizontal gene transfer may skew the topology of the phylogenetic tree constructed based on limited genes, leading to misrepresentation of the phylogenetic relationships. Therefore, a genomewide phylogenetic analysis was conducted in the present study. Multiple sequence comparisons among the significant genera within Trichosporonaceae confirmed that the isolated fungus was A. cacaoliposimilis. Genome homologous family analysis and genome synteny analysis also corroborated this finding.
Biochemical analyses are often used as a crucial tool in fungal identification. However, these analyses are not commonly used as routine clinical tests due to their low accuracy and limited clinical benefit. In the present study, the biochemical reactions of the clinical isolates A. cacaoliposimilis UB0827 and A. mycotoxinivorans GMU 1709 (Peng et al. 2019) were compared with those of the environmental isolate A. cacaoliposimilis ATCC® 20505™ (Gujjari et al. 2011). The differential results obtained are presented in Table I. Certain assimilations, including glycerol, D-melibiose, D-melezitose, and sorbose assimilation, were weaker in the two clinical isolates than those in the environmental isolated strain ATCC® 20505™. These observations, together with the annotations from the CAZy database, revealed that the UB0827 strain had non-significantly higher levels of glycosyltransferases than those noted in GMU1709.
Further, the clinical isolates did not produce β-N-acetylglucosaminidase, while the environmental isolate was positive in the D-glucosamine reaction test. Both the clinical and environmental isolates exhibited a rough phenotype. These findings contrast with those reported by Ichikawa et al. (2004), suggesting that the selective evolution has been possibly influenced by the absence of glucose in the urine. Consistent with characteristics typical of the Trichosporon genus, A. cacaoliposimilis produces urease (Ashok et al. 2019), which is a distinguishing feature for identifying the fungi belonging to this group.
Clinical research on the species related closely to the Trichosporon genus has revealed that these fungi are opportunistic pathogens, particularly in patients admitted to the intensive care units and undergoing invasive procedures and treatment with broad-spectrum antibiotics (Ahangarkani et al. 2021). This represents a significant challenge for the future applications of food additives. Epidemiological studies conducted in Brazil and China have indicated a predominance of infections caused by T. asahii cultured from the samples collected from the urinary tract (de Almeida et al. 2017; Francisco et al. 2019). Since the Apiotrichum genus reclassified in 2015 (Liu et al. 2015b), its clinical pathogenicity has been increasingly recognized. Lara et al. (2019) isolated Apiotrichum veenhuisii from the skin of pediatric patients with acute myeloid leukemia. Peng et al. (2019) reported the presence of A. mycotoxinovorans in the sputum of a pediatric patient with congenital heart disease and pneumonia. Noguchi et al. (2020) isolated A. cacaoliposimilis from the fingernails of a geriatric patient with interstitial pneumonia. These findings indicate the pathogenic potential of the Apiotrichum genus. In the present study, A. cacaoliposimilis UB0827 was isolated from the clear mid-stream urine of a patient with UTI who had undergone invasive catheterization and exhibited improvement after antifungal treatment. The present study revealed intergeneric variations in the epimerase gene (PHI: 5267), which is involved in the synthesis of galactosaminogalactan, a substance capable of modulating host immune response and enhancing pathogenicity (Lee et al. 2015).
Similarly, the gene encoding phosphatidyl-N-(methylethanolamine) N-methyltransferase (PHI: 8896), which is involved in the CDP-DAG pathway, was revealed to be functional in UB0827 (Tams et al. 2019). The CDP-DAG pathway is significant for synthesizing critical phosphatidic acids, such as phosphatidylserine, phosphatidylethanolamine, and phosphatidylcholine in Candida albicans. These phospholipids are essential components of the fungal cell membrane and play critical roles in maintaining cell structure, signaling, and membrane trafficking. The gene encoding serine/threonine-protein kinase (PHI: 9135) is primarily involved in the formation and degradation of autophagosomes, particularly those in the kidney (Ding et al. 2018).
Further, the present study identified cdc4 (PHI: 11985), which encodes a member of the F-box protein family, in the isolated fungus. This gene plays a crucial role in several biological processes, including the cell cycle and fungal infectivity, as the protein encoded by this gene recognizes and ubiquitinates substrates via the ubiquitin ligase E3 complex (Wu et al. 2022). In the pathogenicity syntenic analysis of the clinical strain GMU1709, several independent pathogenicity genes were identified initially, although a few of these genes had to be excluded due to the stringent comparison model. Ultimately, only the high virulence genes aspB and aspC, and the lethal factors pmt4, pri1, met2, and pmt1 that belonged to UB0827 alone, were retained. The genes aspB and aspC, mainly are involved in synthesizing septins, a group of GTPases crucial for maintaining the integrity of the fungal cell wall. The absence of septins could compromise the invasive capabilities of a fungus (Vargas-Muñiz et al. 2015). These findings suggested a direct link between the identified causative genes and the opportunistic infections caused by A. cacaoliposimilis. However, since this strain is the first to be identified and has its whole-genome sequencing data unavailable, intraspecies comparisons could not be conducted at present, and classification based on the clinical origin of the strain did not yield significant results.
The in vitro drug sensitivity assessments revealed that A. cacaoliposimilis was particularly susceptible to triazole antifungal medications, demonstrating the highest sensitivity to voriconazole and posaconazole. Itraconazole was also effective against A. cacaoliposimilis, while fluconazole exhibited a relatively lower activity against this fungus. These findings are consistent with the drug sensitivity statistics reported for the Trichosporon genus in recent years (Francisco et al. 2019; Ahangarkani et al. 2021). However, A. mycotoxinovorans GMU 1709 exhibited less sensitivity to voriconazole compared to itraconazole (Peng et al. 2019), and the literature contains reports of the latter being used prophylactically for T. asahii infections (Asada et al. 2006). In contrast to A. mycotoxinovorans, A. cacaoliposimilis exhibited sensitivity to 5-fluorocytosine. Amphotericin-B was highly effective against A. cacaoliposimilis, while anidulafungin and micafungin exhibited reduced activity, consistent with the previous reports on A. veenhuisii (Lara et al. 2019). Moreover, a comparison with the Comprehensive Antibiotic Resistance Database revealed abundant primary resistance mechanisms, such as efflux, reduced antibiotic permeability, and target alteration-related proteins, in UB0827. These mechanisms are the primary factors underlying echinocandin resistance in the related Apiotrichum spp. (Franconi and Lupetti 2023). When selecting antifungal agents for patients with renal impairments or those at risk of this condition, it is important to consider the pharmacokinetic properties and nephrotoxic potential of such agents to ensure the safety and effectiveness of prophylaxis or treatment (Tragiannidis et al. 2021). Therefore, empirical antifungal therapy alone is inadequate, and routine drug susceptibility testing is necessary.
In summary, infections with Apiotrichum spp. are relatively rare and reported primarily in cutaneous infections. The urinary tract infections (UTIs) involving this genus are usually associated with invasive procedures such as catheterization, which may facilitate fungal colonization. The present study pioneers in reporting the case of an A. cacaoliposimilis strain isolated from the urine sample of a patient with UTI. The present study’s findings are valuable for understanding the clinical characteristics of this fungus. Triazole antibiotics are used empirically for the treatment of A. cacaoliposimilis infections. The subsequent genome-wide analysis classified A. cacaoliposimilis within the Trichosporonaceae family. Identifying distinct inter-generic virulence genes supported the independent positioning of the Apiotrichum genus from the Trichosporon genus in the evolutionary setup. In addition to underscoring the taxonomic separation, the differentiation provides insights for future research on the possible pathogenic mechanisms of these genera.