
Weinfehler spielen in der Analytik schon seit geraumer Zeit eine wichtige Rolle, um allfällige sensorische Beanstandungen quantitativ besser nachweisen zu können. Auf Grund der Vielzahl von Fehlern hat sich hier ein umfangreiches Forschungsgebiet etabliert. Wie auch in anderen Untersuchungsmethoden sind hier vor allem schnelle, kostengünstige und mit einem geringen Volumen an eingesetzter Analysensubstanz verbundenen Techniken gefragt. Als eine der erfolgreichsten Methoden hat sich hier die SPME-Technik etabliert. Für die Korrektur des hier häufig auftretenden Matrix-Effektes ist allerdings wie bei Brandes et al. 2023 erläutert, die Verwendung von internen Standards der entsprechenden Zielsubstanzen in zumindest teilweise isotopenmarkierter Form notwendig. Diese nicht immer kommerziell erhältlichen internen Standards können in einigen Fällen durch Eigensynthese hergestellt werden.
2-Aminoacetophenon gilt als Leitsubstanz für den als „Untypische Alterungsnote“ bezeichneten Weinfehler. Die Bildung in Wein aus dem Precursor Indolessigsäure wurde in mehreren Arbeiten weitgehend geklärt (Christoph et al., 1996; Christoph et al., 1998; Hoenicke et al., 2002; Mihaljecic et al., 2015). 2-AAP ist in 5-fach deuterierter Form kommerziell von der Fa. Eptes-Food&Flavours Analytical erhältlich. Bei dieser Verbindung sind drei Deuteriumatome an der Methylgruppe lokalisiert, während die restliche zwei in ortho- und para-Stellung zur Aminogruppe direkt am aromatischen Kern gebunden sind. Bei den Deuteriumatomen der Methylgruppe ist in wässriger Lösung auf Grund der Keto-Enol-Tautomerie allerdings ein Austausch durch Wasserstoffatome zu erwarten. 2-AAP ist zwar in unverändertem Zustand der SPME-Analytik zugänglich (Fan et al., 2007; Mihaljevic et al.; 2015, Prudencio et al 2018; Schmarr et al., 2016), jedoch sind Selektivität und Sensitivität durch Derivatisierung noch beträchtlich steigerbar (Brandes et al., 2016).
2,4,6-Tribromanisol und 2-Methylisoborneol sind neben einer Reihe anderer Substanzen (2,4,6- Trichloranisol, Geosmin, u.a.) mögliche Verursacher für die als korkig bzw. erdig, muffig oder schimmelig bezeichneten Weinfehler. Während Tribromanisol von Mikroorganismen durch Methylierung aus dem Precursor Tribromphenol gebildet wird, ist 2-Methylisoborneol ein Stoffwechselprodukt einer Reihe von Streptomyceten, Pilzen und Cyanobakterien (Pinar et al., 2016; Nauer et al., 2022; Cravero et al., 2020). Obwohl 2,4,6-TBA in volldeuterierter Form mittlerweile von einer Reihe von Firmen (Fisher Scientific, VWR, Toronto Research Chemicals, HPC u.a.) kommerziell erhältlich ist, war zu Beginn der Planung dieser Arbeit diese Substanz lediglich aus Übersee zu kaufen. Während der Synthesearbeiten erfolgte dann eine wesentliche Erweiterung des Angebotes auch im europäischen Raum. Die Synthese wurde dennoch fortgesetzt, vor allem um Erfahrungen bezüglich Möglichkeiten und Schwierigkeiten bei der Herstellung deuterierter aromatischer Systeme zu gewinnen.
Bei 2-MIB ist lediglich die Verbindung mit drei Deuteriumatomen an der Methylgruppe des Kohlenstoffatoms 2 erhältlich (Sigma Aldrich, Santa Cruz Biotechnologie, Toronto Research Chemicals, Neochema u.a.). Beide Verbindungen sind ohne weitere Derivatisierung mittels SPME-Technik mit ausgezeichneter Empfindlichkeit detektierbar (Arrebola-Liebianas et al., 2016; Canpillo et al., 2008; Chatonnet et al., 2004, Jönsson et al., 2006; Lopez et al., 2009; Martendal et al., 2007; Ligor et al., 2006). Die Erfassung von 2-MIB-d3 mit Elektronenstoßionisation und nachfolgender massenspektrometrischer Detektion gestaltet sich allerdings, wie weiter unten beschrieben, als ungünstig.
Ziel dieser Arbeit war eine mögliche Erleichterung der Messmethodik für die Weinfehltöne „Untypische Alterungsnote“ (2-AAP) und #x201E;Kork-Schimmelton“ (2-MIB, 2,4,6-TBA) durch die Synthese der Verbindungen 2-Aminoacetophenon-d4, 2,4,6-Tribromanisol-d5 und 2-Methylisoborneol-d7 als interne Standards für die SPME-Analytik.
Aluminiumchlorid wasserfrei 98 % (Roth, Karlsruhe, BRD)
Dichlormethan 99,5 % (Roth, Karlsruhe, BRD)
Benzol-d6 99,5 Atom % (Roth, Karlsruhe, BRD)
Acetylchlorid 99 % (Sigma Aldrich, St. Louis, Missouri, USA)
Salzsäure p.A. 37 % (Roth, Karlsruhe, BRD)
Natriumsulfat wasserfrei gekörnt 99,5 % (Roth, Karlsruhe, BRD)
Salpetersäure 99,5 % (Sigma Aldrich, St. Louis, Missouri, USA)
Natriumhydroxid p.A. 97 % (Roth, Karlsruhe, BRD)
tert-Butylmethylether 99,5 % (Roth, Karlsruhe, BRD)
Eisenpulver 99,5 % (Roth, Karlsruhe, BRD)
Ethanol-d1 99 Atom % (Sigma Aldrich, St. Louis, Missouri, USA)
Deuteriumchlorid 35 % in D2O 99 Atom % (Sigma Aldrich, St. Louis, Missouri, USA)
Natriumacetat p.A. 99 % (Roth, Karlsruhe, BRD)
Essigsäure p.A.100 % (Roth, Karlsruhe, BRD)
Kaliumhydrogencarbonat 99,5 % (Roth, Karlsruhe, BRD)
Natriumchlorid (Sigma Aldrich, St. Louis, Missouri, USA)
Ethanol 100 % (Roth, Karlsruhe, BRD)
Schwefelsäure 96 % (Roth, Karlsruhe, BRD)
Natriumcarbonat 99 % (Roth, Karlsruhe, BRD)
Calciumchlorid 96 % granuliert (Roth, Karlsruhe, BRD)
Hydrazinmonohydrat 64-65 % (Sigma Aldrich, St. Louis, Missouri, USA)
Aluminium-Nickel-Legierung (Sigma Aldrich, St. Louis, Missouri, USA)
Deuteriumoxid 99,9 Atom % (Sigma Aldrich, St. Louis, Missouri, USA)
Schwefelsäure-d2 98 % 99,5 Atom % (Roth, Karlsruhe, BRD)
Natriumnitrit 99 % (Roth, Karlsruhe, BRD)
Natriumhydroxid-d 99 Atom %, 40%ige Lösung in D2O (Sigma Aldrich, St. Louis, Missouri, USA)
Natriumdithionit techn (Sigma Aldrich, St. Louis, Missouri, USA)
Brom 99,5 % (Sigma Aldrich, St. Louis, Missouri, USA)
Kaliumdisulfit 98 % (Sigma Aldrich, St. Louis, Missouri, USA)
Phosphorpentoxid 99 % (Roth, Karlsruhe, BRD)
Methanol-d1 99 Atom % (Sigma Aldrich, St. Louis, Missouri, USA)
Iodmethan-d3 99,5 Atom % (Roth, Karlsruhe, BRD)
Isohexan 99 % (Roth, Karlsruhe, BRD)
(-) Bornylacetat 95 % (Sigma Aldrich, St. Louis, Missouri, USA)
Chrom(VI)oxid p.S. (Sigma Aldrich, St. Louis, Missouri, USA)
Methanol 99,5 % (Roth, Karlsruhe, BRD)
Cyclohexan 99,5 % (Roth, Karlsruhe, BRD)
Diethylenglykol p.S. 99 % (Roth, Karlsruhe, BRD)
Natrium 99 % (Roth, Karlsruhe, BRD)
Hydrazin-d4 Monodeuterated (Sigma Aldrich, St. Louis, Missouri, USA)
Natriumhypochloritlösung 12 % C, techn. (Roth, Karlsruhe, BRD)
Lanthan(III)-Chlorid-Bis(lithiumchlorid)-Komplex-Lösung (Sigma Aldrich, St. Louis, Missouri, USA)
Tetrahydrofuran 99,5 % (Roth, Karlsruhe, BRD)
Ammoniumchlorid 99 % (Roth, Karlsruhe, BRD)
Magnesium Späne p.S. 99,9 % (Roth, Karlsruhe, BRD)
Diethylether p.S. 99,5 % (Roth, Karlsruhe, BRD)
Petrolether 40-60°C Rotisolv HPLC (Roth, Karlsruhe, BRD)
Kieselgel 60 (Roth, Karlsruhe, BRD)
Gaschromatograph: 6890N Gaschromatograph mit 5975 massenspektrometrischem Detektor (beides Fa. Agilent, St. Clara, USA)
Trennsäule: DB-5 60m/0,25 ID/0,25μm Film (Fa. Agilent, St. Clara, USA)
Gaschromatograph: 7820A mit Flammenionisationsdetektor (beides Fa. Agilent, St. Clara, USA)
Trennsäule: DB-5 60m/0,32 ID/0,25μm Film (Fa. Agilent, St. Clara, USA)
Die Gehaltsbestimmung der Endprodukte erfolgte gaschromatographisch durch Flächenvergleich mit den nicht deuterierten Substanzen. Die Massenspektren wurden auf einem Gaschromatographen mit massenspektrometrischer Detektion aufgenommen.
15 g AlCl3 wasserfrei werden mit 15 ml trockenem Dichlormethan und 9,6 ml Benzol-d6 unter Rühren gemischt. Zu der Mischung werden langsam unter Rühren und Eiskühlung 7,8 ml Acetylchlorid zugetropft. Die Reaktion muss wegen der dabei erfolgenden Deuteriumchlorid-Entwicklung in einem Abzug durchgeführt werden. Die Mischung wird nach Beendigung der Zugabe noch 30 min bei Raumtemperatur gerührt und über Nacht stehen gelassen. Das Reaktionsgemisch wird unter Rühren langsam auf 100 ml Eis gegossen und eine etwaige Trübung durch Zugabe von wenig 10%iger HCl beseitigt. Die Lösung wird in einen Scheidetrichter überführt, die organische Phase abgetrennt und die wässrige Phase noch dreimal mit je 10 ml Dichlormethan extrahiert. Die vereinigten Extrakte werden mit Natriumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer weitgehend entfernt. Das Rohprodukt wird durch Destillation bei ca. 10 mbar gereinigt.
In 40 ml 99,5%ige Salpetersäure werden bei -10 °C 6 g Acetophenon-d5 unter Rühren so langsam zugetropft, dass die Temperatur nicht über -8 °C steigt und anschließend noch 15 min stehen gelassen. Das Reaktionsgemisch wird in 400 ml Eiswasser gegossen und unter weiterer Kühlung mit 20%iger Natronlauge alkalisch gemacht. Danach wird die Lösung in einen Scheidetrichter überführt und viermal mit je 50 ml tert-Butylmethylether extrahiert. Die vereinigten Extrakte werden mit Natriumsulfat getrocknet. Das Lösungsmittel wird am Rotationsverdampfer bei einer Heizbadtemperatur von 50 °C bis zur Abscheidung von Kristallen eingedampft und die verbleibende Lösung im Kühlschrank bei 4 °C über Nacht stehen gelassen. Die Flüssigkeit wird von den abgeschiedenen Kristallen vorsichtig abgehoben, am Rotationsverdampfer bei einer Heizbadtemperatur von 50 °C auf das ca. halbe Volumen eingedampft und anschließend wieder über Nacht im Kühlschrank bei 4 °C stehen gelassen. Dieser Vorgang wird bis zu einem Restvolumen von ca. 5 ml wiederholt, das Lösungsmittel anschließend vollständig entfernt und das Rohprodukt bei ca. 0,5 mbar destilliert. Das ölige, hellgelbe Endprodukt besteht durchschnittlich aus ca. 75 % 2-Nitroacetophenon-d4, 20 % 3-Nitroacetophenon-d4 und 5 % anderen Nebenprodukten.
2 g Nitroacetophenon-d4 (Syntheseprodukt) werden nach Zugabe von 2,54 g Eisenpulver in 20 ml Ethanol-d1 gelöst. Die Reaktionsmischung wird auf ca. 60 °C erwärmt und anschließend unter Rühren 9 ml 36%ige Deuteriumchloridlösung tropfenweise zugegeben. Nach der letzten Zugabe wird die Mischung 1 Stunde unter Rückfluss zum Sieden erhitzt. Die abgekühlte Reaktionsmischung wird unter Rühren in ein Gemisch aus 300 ml 4%ige Natriumacetatlösung und 1 ml Essigsäure gegossen, in einen Scheidetrichter überführt und viermal mit je 25 ml t-Butylmethylether extrahiert. Die vereinigten Extrakte werden mit 20 ml gesättigter Kaliumhydrogencarbonatlösung säurefrei gewaschen und mit Natriumsulfat getrocknet. Nach Entfernung des Lösungsmittels am Rotavapor wird der Rückstand direkt einer Wasserdampfdestillation unterworfen. Nach ca. 100 ml Destillat wird abgebrochen, das Destillat mit 15 g NaCl versetzt und bis zur völligen Auflösung gerührt. Die Lösung wird in einen Scheidetrichter überführt und viermal mit je 10 ml Dichlormethan extrahiert. Die Extrakte werden mit Natriumsulfat getrocknet und anschließend das Lösungsmittel am Rotationsverdampfer entfernt. Der Rückstand wird in 20 ml 50%igem Ethanol gelöst, mit 50 μl 2molarer Natronlauge versetzt und eine Stunde unter Rückfluss zum Sieden erhitzt. Nach Abkühlen werden ca. 70 ml Deionat zugegeben, in einen Scheidetrichter überführt und 3mal mit je 25 ml Dichlormethan extrahiert. Die vereinigten Extrakte werden mit Natriumsulfat getrocknet und das Lösungsmittel anschließend am Rotavapor entfernt. Der Rückstand wird bei ca. 0,5 mbar destilliert. Das Endprodukt ist ca. 98%ig.
Zu 6,1 ml Benzol-d6 werden im Eisbad unter intensivem Rühren ein Gemisch von 3,8 ml 99,5%ige Salpetersäure und 4,6 ml 96%ige Schwefelsäure langsam zugetropft, so dass die Temperatur 5 °C nicht übersteigt. Nach Beendigung der Zugabe wird das Reaktionsgemisch unter weiterem Rühren im Wasserbad 2 Stunden auf ca. 55 °C erhitzt. Das abgekühlte Gemisch wird in ein Zentrifugenröhrchen überführt und 5min bei 6000 U/min zentrifugiert. Das aufschwimmende Nitrobenzol-d5 wird vorsichtig abgehoben, mit 5 ml 10%iger Natriumcarbonatlösung versetzt, intensiv gemischt und wie oben zentrifugiert. Die obere, wässrige Phase wird verworfen und das verbleibende Nitrobenzol-d5 noch zweimal mit je 5 ml Deionat gewaschen. Nach dem letzten Waschschritt wird das Rohprodukt mit Calciumchlorid getrocknet und durch Vakuumdestillation bei ca. 10 mbar gereinigt.
Das erhaltene Nitrobenzol-d5 wird in Ethanol gelöst (10 ml Ethanol/g Nitrobenzol), mit Hydrazinhydrat gemischt (1,25ml Hydrazinhydrat 65 % pro ml Nitrobenzol) und auf ca. 40 °C erwärmt. Nach Zugabe von ca. 100mg frisch präpariertem Raney-Nickel wird der Kolben bis zur Beendigung der Reaktion (Lösung fast farblos, nur mehr geringe Gasentwicklung) an einen Rückflusskühler angeschlossen. Danach werden nochmals 200 mg Raney-Nickel zugegeben und unter Rückfluss 1 Stunde gekocht. Nach Abkühlen wird die Lösung am Rotationsverdampfer bis zur beginnenden Trübung eingeengt und anschließend direkt einer Wasserdampfdestillation unterworfen. Nach 70 ml Destillat wird abgebrochen, das Destillat mit 15 g Natriumchlorid versetzt und bis zur völligen Auflösung gerührt. Die Lösung wird in einen Scheidetrichter überführt und 4 mal mit je 10 ml Dichlormethan extrahiert. Die Extrakte werden mit Natriumsulfat getrocknet und anschließend das Lösungsmittel am Rotationsverdampfer weitgehend entfernt.
Das erhaltene Anilin-d5 wird zu einer Mischung aus Deuteriumoxid und Schwefelsäure-d2 (5 ml Deuteriumoxid und 1,4 ml Schwefelsäure-d2 96 % pro ml Anilin) unter Rühren zugegeben und das Gemisch anschließend im Eisbad gekühlt. Danach wird tropfenweise unter intensivem Rühren eine Lösung von Natriumnitrit in Deuteriumoxid (0,72 g Natriumnitrit in 1 ml Deuteriumoxid pro ml Anilin) zugegeben und nach Beendigung der Zugabe 30 min lang auf ca. 80 °C erhitzt. Die Lösung wird anschließend wasserdampfdestilliert und ca. 20 ml Wasserdampf pro ml eingesetztes Anilin-d5 abdestilliert. Das Destillat wird mit Kochsalz gesättigt, in einen Scheidetrichter überführt und 4 mal mit je 10 ml Dichlormethan extrahiert. Die Extrakte werden mit Natriumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer weitgehend entfernt. Das erhaltene, leicht bräunliche Rohphenol wird in Deuteriumoxid gelöst (1 ml Deuteriumoxid pro ml Phenol), und die Lösung durch Zusatz von Natriumdeuteroxid alkalisch gemacht (0,25 ml Natriumdeuteroxid 40 % pro ml Phenol). Die Lösung wird auf etwa 70 °C erwärmt und anschließend so lange Natriumdithionit in kleinen Mengen unter Schütteln zugegeben, bis Entfärbung eintritt. Die noch warme Lösung wird in 50 ml 1%ige Salzsäure pipettiert, in einen Scheidetrichter überführt und 4 mal mit je 10 ml Dichlormethan extrahiert. Die Extrakte werden mit Natriumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das erhaltene, leicht gelbliche Phenol-d5 wird im Vakuum bei ca. 10 mbar destilliert.
1ml Phenol-d5 werden in 70 ml Deionat gelöst. Anschließend werden langsam und unter Rühren 16 ml einer 2molaren Bromlösung zu Essigsäure zugegegeben und 30 min gerührt. Danach werden 0,2 g Kaliumdisulfit zugegeben und weitere 30 min gerührt. Anschließend wird das ausgefallene 2,4,6-TBA-d2 durch eine Nutsche abfiltriert, mit Deionat gewaschen und weitere 30 min trocken gesaugt. Der noch feuchte Niederschlag wird in einen 100 ml Rundkolben überführt und mit 40%- igem Ethanol umkristallisiert. Der Niederschlag wird wie oben abgesaugt, mit wenig kaltem 40%- igem Ethanol gewaschen, trockengesaugt und danach im Exsikkator zuerst über Kieselgel und danach über Phosphorpentoxid getrocknet.
1g 2,4,6-TBP-d2 werden in 10 ml Methanol-d1 gelöst. Nach Zugabe von 0,22ml Natronlauge-d1 40 % und 0,21 ml Iodmethan-d3 wird gemischt und drei Stunden unter Rückfluss zum Sieden erhitzt. Die noch warme Lösung wird in einen Scheidetrichter mit 100 ml 0,5molare Natronlauge überführt, mit 50 ml Isohexan extrahiert und die wässrige Phase nach der Phasentrennung verworfen. Die Isohexanphase wird mit je 50 ml 0,5molare Natronlauge so lange gewaschen, bis kein 2,4,6-TBP-d2 mehr nachweisbar ist. Dann wird das Isohexan mit Natriumsulfat getrocknet, das Lösungsmittel am Rotationsverdampfer entfernt und der Rückstand im Vakuum einer Membranpumpe getrocknet. Das Endprodukt ist ca. 98%ig.
Pro Ansatz werden 2 g Bornylacetat in 30 ml Essigsäure gelöst und unter Rückfluss und Rühren zum Sieden erhitzt. Anschließend werden über einen Zeitraum von drei Stunden insgesamt 5 g Chromtrioxid in kleinen Portionen durch den Rückflusskühler zugegeben und nach der letzten Zugabe noch eine Stunde unter Rückfluss zum Sieden erhitzt. Nach dem Abkühlen wird mit 140 ml Deionat verdünnt, in einen Scheidetrichter überführt und 3 mal mit je 25 ml Dichlormethan extrahiert. Die vereinigten Extrakte werden mit gesättigter Kaliumhydrogencarbonatlösung säurefrei gewaschen und anschließend mit Natriumsulfat getrocknet. Das Lösungsmittel wird am Rotavapor entfernt. Die Rückstände mehrerer Ansätze werden über eine 10 cm lange Vigreuxkolonne bei 0,1 mbar in eine leichter und schwerer flüchtige Fraktion getrennt. 2 g schwer flüchtige Fraktion wird mit 1,1 g Natriumcarbonat, 8 ml Methanol und 16 ml Deionat eine Stunde unter Rückfluss zum Sieden erhitzt und über Nacht bei Raumtemperatur stehen gelassen. Die Lösung wird danach in einen Scheidetrichter überführt, mit 30 ml Deionat verdünnt und 3 mal mit je 25 ml Dichlormethan extrahiert. Die organischen Phasen werden mit Natriumsulfat getrocknet und das Lösungsmittel am Rotavapor entfernt. Der Rückstand wird mit Cyclohexan umkristallisiert und ergibt ca. 90%iges 5-Oxoborneol. Auch durch weiteres Umkristallisieren konnte der Reinheitsgrad nicht wesentlich erhöht werden, so dass mit dem 90%igem Produkt weitergearbeitet wurde.
Pro Ansatz werden 540 mg 5-Oxoborneol in 6 ml Methanol-dl gelöst. Nach Zugabe von 20 μl Natriumdeuteroxidlösung (40 %) wird unter Rühren eine Stunde unter Rückfluss zum Sieden erhitzt. Nach dem Abkühlen wird das Lösungsmittel im Vakuum einer Membranpumpe möglichst vollständig entfernt. Der Rückstand wird mit 8 ml Diethylenglykol-d2, in dem vorher ca. 300 mg Natrium unter Erwärmen gelöst wurden, versetzt und nach Zugabe von 0,45 ml Hydrazin-d4 monodeuterat 4 Stunden unter Rückfluss auf ca. 190 °C erhitzt. Nach dem Abkühlen wird mit 8 ml Deionat verdünnt und anschließend 2 mal mit je 10 ml tertButylmethylether extrahiert. Die vereinigten Extrakte werden 2 mal mit je 10 ml Deionat gewaschen, mit Natriumsulfat getrocknet und das Lösungsmittel am Rotavapor entfernt. Das leicht bräunliche Rohprodukt wird bei 8 mbar und 100 °C sublimiert.
Für die Herstellung von Diethylenglykol-d2 werden 26 ml Diethylenglykol mit 10 ml Deuteriumoxid 1 Stunde unter Rückfluss und Ausschluss von Feuchtigkeit zum Sieden erhitzt. Nach dem Abkühlen wird das Wasser im Vakuum einer
Membranpumpe möglichst vollständig abdestilliert. Der Prozess wird noch 3 mal wiederholt und zum Schluss das zurückbleibende Diethylenglykol-d2 im Vakuum bei 1 mbar destilliert und das Destillat in einem Exsikkator über Phosphorpentoxid aufbewahrt.
500 mg Borneol-d4 werden in 5ml Eisessig gelöst. Unter Rühren werden innerhalb einer Stunde 3 ml Natriumhypochloritlösung (1,2 molar) in Portionen von jeweils 0,2 ml zugesetzt. Die Lösung wird dabei durch Kühlen auf einer Temperatur zwischen 15 und 25 °C gehalten. Nach der letzten Zugabe wird mit Kaliumiodid-Stärkepapier auf freies Hypochlorit geprüft, wenn nötig weitere Hypochloritlösung zugegeben und anschließend 1 Stunde bei Raumtemperatur gerührt.
Danach wird so lange gesättigte Kaliumdisulfitlösung zugesetzt, bis die gelbe Farbe verschwindet und die Reaktion mit Kaliumiodid-Stärkepapier negativ ist. Die Lösung wird mit 50 ml Deionat verdünnt und anschließend 3 mal mit je 10 ml Dichlormethan extrahiert. Die vereinigten Extrakte werden mit gesättigter Natriumhydrogencarbonatlösung säurefrei gewaschen, mit Natriumsulfat getrocknet und das Lösungsmittel am Rotavapor entfernt. Das leicht gelbliche Rohprodukt wird bei 8 mbar und 100 °C sublimiert.
620 mg Kampher-d4 werden in einer Mischung aus 9 ml trockenem Tetrahydrofuran und 3,2 ml Lanthan(III)Chlorid-Bis(lithiumchlorid)-Komplex – Lösung eine Stunde bei Raumtemperatur unter Feuchtigkeitsausschluss gerührt. Danach wird im Eisbad gekühlt und mit Stickstoff gespült. Anschließend werden unter Rühren 3 ml einer ca. 2molaren Lösung von Trideuteromethylmagnesiumiodidlösung tropfenweise zugegeben. Nach Ende der Zugabe wird das Eisbad entfernt und die Lösung noch 3 Stunden bei Raumtemperatur gerührt. Danach wird wieder im Eisbad gekühlt und langsam und unter Rühren 15 ml gesättigte Ammoniumchloridlösung zugegeben. Die Suspension wird noch 10 Minuten gerührt, danach in ein Zentrifugenröhrchen überführt und 2 Minuten bei 4000U/min zentrifugiert. Der Überstand wird abgehoben und die verbleibende wässrige Phase noch einmal mit 10 ml tert-Butylmethylether extrahiert. Die vereinigten organischen Phasen werden mit Natriumsulfat getrocknet und das Lösungsmittel anschließend am Rotavapor entfernt. Der Rückstand wird im Vakuum bei ca. 10 mbar und 100 °C sublimiert. Das Rohprodukt wird auf einer Kieselgelsäule mit einem Gemisch von 5 Vol% tert-Butylmethylether in Petrolether als Laufmittel gereinigt. Das Endprodukt ist ca. 94%ig.
Für die Herstellung des Grignardreagenzes werden 300 mg Magnesium in einem trockenen, mit Stickstoff gespülten 25ml Zweihalskolben mit Rührstab mit 4 ml trockenem Diethylether überschichtet. Anschließend werden 50 μl Iodmethan-d3 zugegebenund der der Kolben in ein Ultraschallbad gestellt, bis eine leichte Trübung sichtbar ist. Danach werden langsam eine Mischung aus 0,7 ml Iodmethan-d3 und 1,5 ml Diethylether zugegeben. Danach wird noch 30 Minuten bei Raumtemperatur gerührt.
Für einen Ersatz durch Deuterium kommen lediglich die 4 Kernwasserstoffe in Frage. Sowohl die beiden Wasserstoffatome der Aminogruppe als auch die 3 Wasserstoffatome der Acylgruppe unterliegen in protischen Lösungsmitteln einem Austausch. Als Ausgangsverbindung wurde Benzol-d6 gewählt. Die Friedl-Crafts-Acylierung mit Acetylchlorid liefert mit sehr guter Ausbeute Acetophenon-d5. Die Nitrierung der Verbindung mit hochkonzentrierter Salpetersäure liefert neben dem Hauptprodukt 3-Nitroacetophenon-d4 eine kleinere Menge an 2-Nitroacetophenon-d4 (Houben-Weyl, 2014). Ein Verlust von Deuterium tritt hierbei nicht ein, da die Nitrierung wesentlich schneller abläuft als der Deuterium-Wasserstoffaustausch und die Nitrogruppe als Substituent 2. Ordnung die Reaktivität bezüglich dieses Austausches stark vermindert. Der Großteil des 3-Nitroacetophenon-d4 sowie anderer, in geringen Mengen gebildeten Nebenprodukte, kann auf Grund der deutlich höheren Schmelzpunkte durch Kristallisation abgetrennt werden. Die Reduktion mit Eisen verläuft selektiv und nahezu quantitativ, muss aber in volldeuterierter Lösung erfolgen, weil 2-Aminoacetophenon im stark sauren Bereich Kerndeuterium austauscht. Für die Aufarbeitung der Lösung vor der Extraktion darf diese weder zu sauer noch zu basisch sein. Im stark sauren Bereich tauscht, wie oben erwähnt, 2-AAP-d4 teilweise Kerndeuterium aus und im alkalischen Bereich fällt Eisenhydroxid aus, das die Extraktion erheblich erschwert. Der Acetatpuffer stellt hier einen guten Kompromiss dar. Die Trennung der im Reaktionsprodukt enthaltenen Verbindungen 2-Aminoacetophenon-d4, 3-Aminoacetophenon-d4 und geringer Mengen anderer Nebenprodukte ist in einfacher Weise durch Wasserdampfdestillation möglich. Hier ist nur 2-AAP-d4 auf Grund der Ausbildung einer intramolekularen Wasserstoffbrücke flüchtig.
Beim Endprodukt müssen die in der Acylgruppe teilweise vorhandenen Deuteriumatome gegen Wasserstoff ausgetauscht werden. Um den Verlust von Kerndeuterium zu vermeiden muss dieser Rücktausch mit einer Base als Katalysator erfolgen. Die Gesamtausbeute an 2-AAP-d4 beträgt bezogen auf die Ausgangsverbindung Benzol-d6 ca. 45 %, wobei die höchsten Verluste bei der Nitrierung von Acetophenon-d5 auftreten, während alle anderen Syntheseschritte mit mehr als 90%-iger Ausbeute stattfinden. Das Massenspektrum zeigt neben dem Molekülion die unter Abspaltung der Methylbzw. Acylgruppe entsprechenden Ionen. Basispeak ist die durch Verlust der Methylgruppe gebildete Struktur (Abb. 1). Entsprechend diesen Ausführungen ist der Massenshift bei dem deuterierten Produkt bei allen drei Hauptionen 4 (Abb. 2), so dass ein größerer Spielraum im Bezug auf die massenspektrometrisch wählbaren Detektionsparameter besteht.
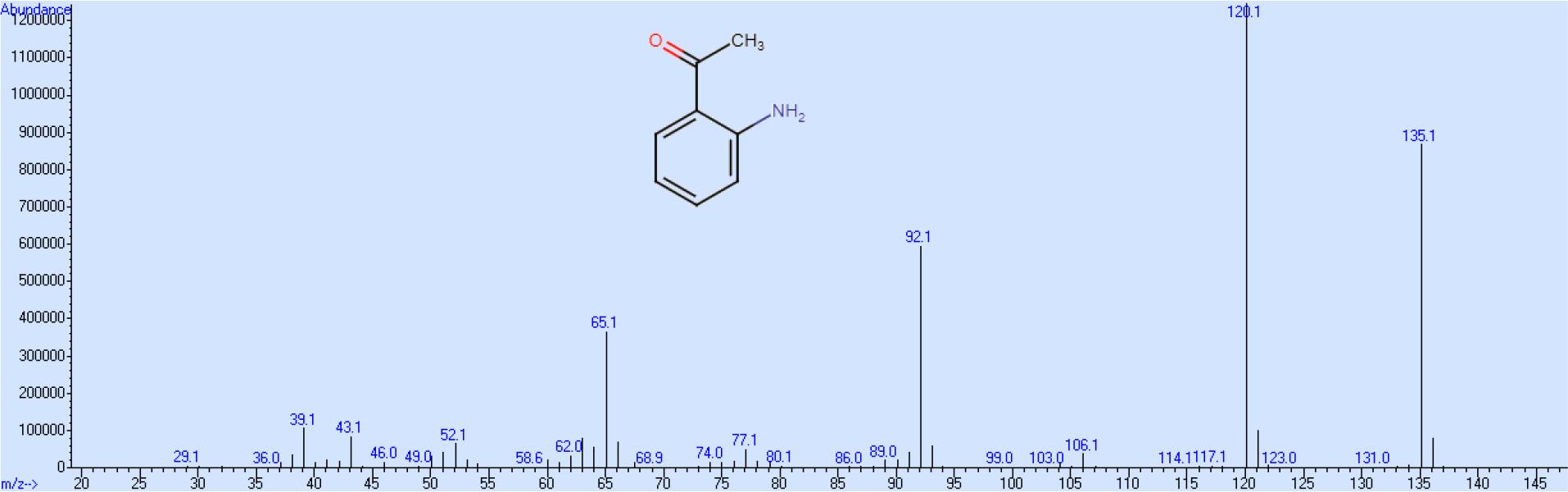
Massenspektrum von 2-Aminoacetophenon
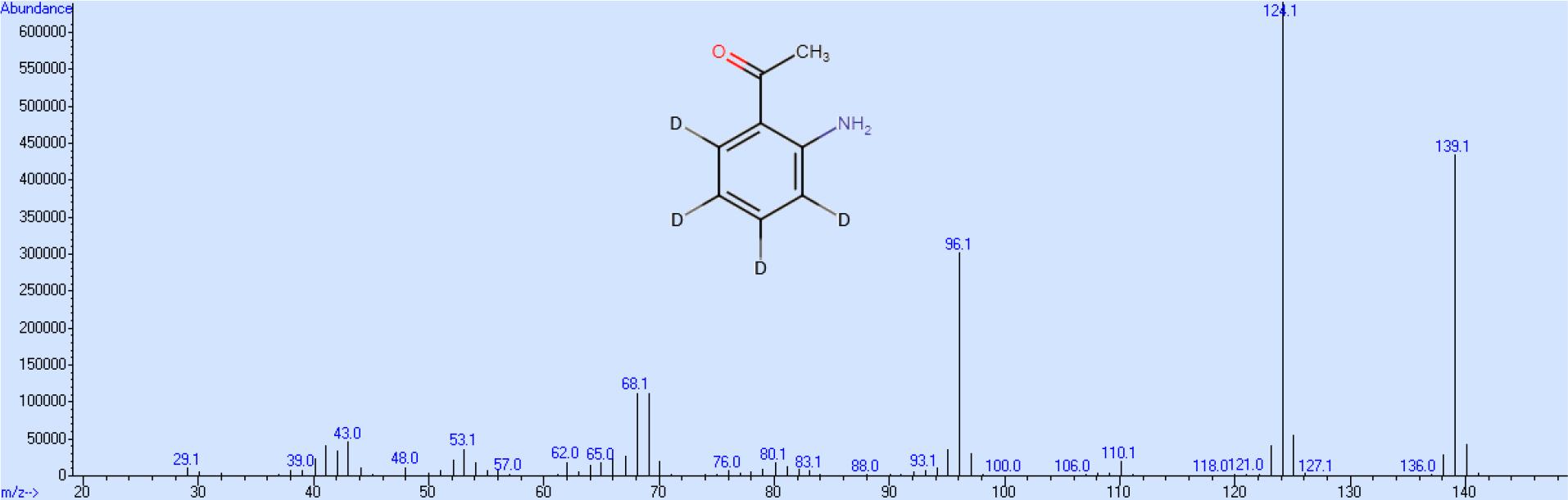
Massenspektrum von 2-Aminoacetophenon-d4
2,4,6-TBA besitzt neben den drei Wasserstoffatomen der Methoxygruppe noch 2 direkt am Kern gebundene. Der Molekülpeak ist gleichzeitig Basispeak. Die Methylgruppe wird unter den Bedingungen der Elektronenstoßionisation in einem ersten Schritt durch alpha-Spaltung abgespalten. Die weitere Elimination von Kohlenmonoxid führt zu einem 5-er Ring. Da Brom zu nahezu gleichen Teilen aus den Isotopen Brom 79 und 81 besteht, treten neben dem Molekülion 344 noch die Massenzahlen 345 bis 349 in nennenswerten Mengen auf. Daraus ergibt sich, dass für ein den Eingangsbedingungen genügendes Produkt, der Ersatz sowohl der beiden am Kern gebundenen Wasserstoffatome als auch die der Methoxygruppe durch Deuterium notwendig ist.
Als Ausgangsverbindung wurde Benzol-d6 gewählt. Die Nitrierung mit einem Gemisch aus Salpetersäure und Schwefelsäure liefert in einem ersten Schritt das entsprechende Nitrobenzol-d5. Ein Verlust von Deuterium tritt hierbei nicht ein, da die Nitrierung wesentlich schneller abläuft als der Deuterium-Wasserstoffaustausch und die Nitrogruppe als Substituent 2. Ordnung die Reaktivität bezüglich dieses Austausches stark vermindert. Die Reduktion des Nitrobenzol-d5 zu Anilin-d5 gelingt mit Hydrazinhydrat und Raney Nickel in ethanolischer Lösung nahezu quantitativ. Auch hier ist kein Verlust an Deuterium zu beobachten, da unter den gewählten Reaktionsbedingungen die Reaktivität des aus dem Hydrazin freigesetzten Wasserstoffs nicht für eine Reaktion mit dem aromatischen Kern ausreicht. Die Umwandlung von Anilin-d5 in Phenol-d5 durch Diazotierung und erwärmen der Diazoniumsalzlösung muss in volldeuterierter Lösung erfolgen, da Anilin bei tiefen pH-Werten sehr leicht Kerndeuterium austauscht. Das erhaltene Phenol-d5 enthält eine kleine Menge 4-Hydroxyazobenzol, dass die weitere Verarbeitung stört und entfernt werden muss. Als einfacher Aufreinigungsschritt bietet sich hier die Spaltung der Azogruppe durch Natriumdithionit in alkalischer Lösung an. Auch dieser Schritt muss in volldeuterierter Lösung erfolgen, da Phenol in alkalischer Lösung leicht Deuterium austauscht.
Die Spaltprodukte der Azoverbindung, Anilin und 4-Aminophenol, besitzen beide basische Aminogruppen und verbleiben bei der nachfolgenden Extraktion unter sauren Bedingungen vorwiegend in der wässrigen Phase. Die verbleibenden Reste an Anilin und 4-Aminophenol stören die weitere Verarbeitung nicht. Die Bromierung von Phenol-d5 zu 2,4,6-Tribromphenol-d2 in wässriger Lösung mit in Eisessig gelöstem Brom erfolgt sehr schnell und nahezu quantitativ. Auch hier tritt kein Austausch von Kerndeuterium ein, da einerseits die Bromierung schneller als der Austausch verläuft und der aromatische Kern durch den starken Elektronenzug der drei Bromatome deaktiviert wird. Das als Nebenprodukt in kleinerer Menge gebildete 2-4,4,6-Tetrabrom-2,5-cyclohexadien-1-on wird durch Kaliumdisulfit quantitativ zu 2,4,6-Tribromphenol reduziert. Als letzter Schritt wird in einer Williamsonsynthese im erhaltenen 2,4,6- Tribromphenol-d2 der Wasserstoff der Hydroxygruppe durch eine volldeuterierte Methylgruppe ersetzt. Nicht umgesetztes 2,4,6-TBP-d2 ist auf Grund seines sauren Charakters vom 2,4,6- TBA-d5 durch wiederholte Extraktion mit verdünnter Natronlauge leicht abtrennbar. Die Gesamtausbeute an 2,4,6-TBA-d5 beträgt bezogen auf die Ausgangsverbindung Benzol-d6 ca. 50 %, wobei die höchsten Verluste bei der Diazotierung von Aminobenzol-d5 und nachfolgender „Verkochung„ der entsprechenden Diazosubstanz zum Phenol-d5 auftreten, während alle anderen Syntheseschritte mit mehr als 90%iger Ausbeute stattfinden. Das Massenspektrum zeigt, wie oben beschrieben, eine Gruppe von Peaks zwischen 342 und 349 als Molekülionen, eine weitere Gruppe zwischen 327 und 333, die dem Verlust der Methylgruppe entsprechen und eine Gruppe zwischen 299 und 306, die dem gebildeten 5-er Ring entsprechen (Abb. 3). Das Massenspektrum der deuterierten Verbindung zeigt, dem Spaltungsmuster entsprechend, bei der ersten Gruppe einen Shift von 5 und bei den anderen beiden Gruppen jeweils einen Shift von 2 (Abb. 4). Dieser Massenshift ist auf Grund des umfangreichen Fragmentierungsmusters von 2-TBA notwendig, um diskriminierende Massen auswählen zu können.
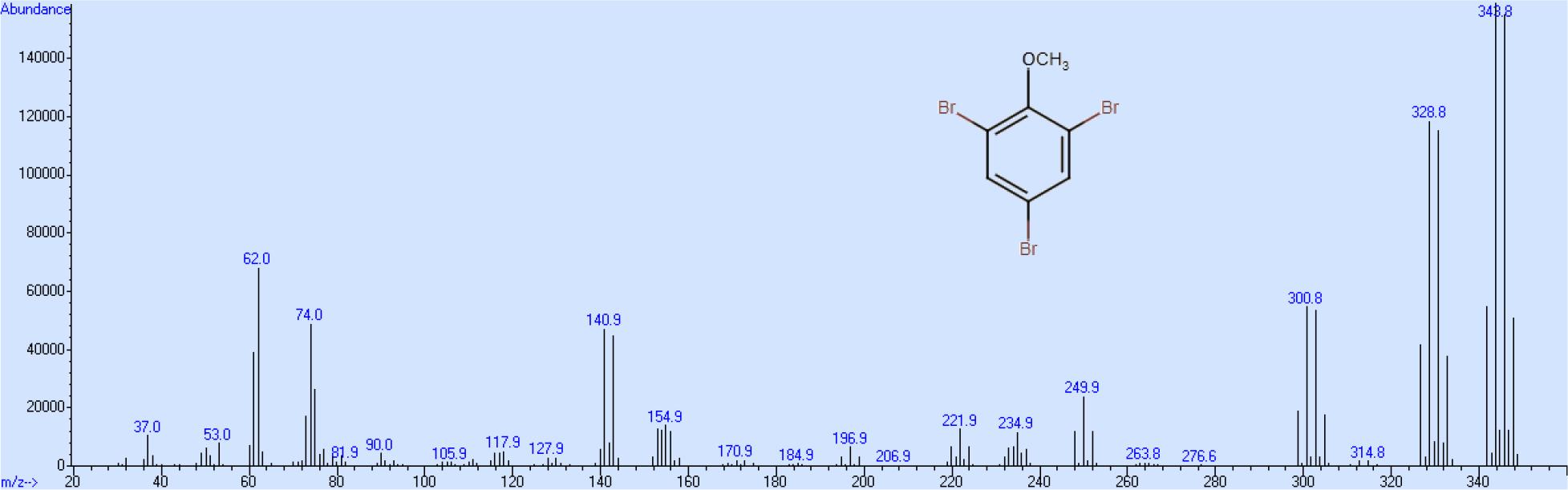
Massenspektrum von 2,4,6-Tribromanisol
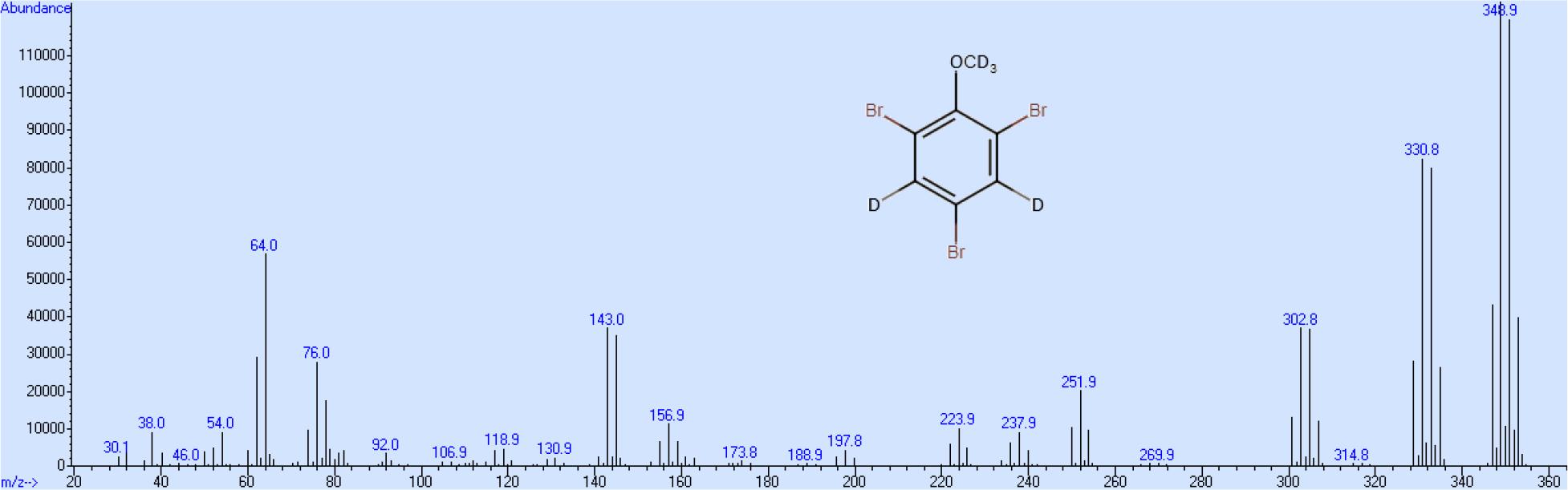
Massenspektrum von 2,4,6-Tribromanisol-d5
Die Fragmentierung von 2-Methylisoborneol unter den Standardbedingungen der Elektronenstoßionisation ergibt als Hauptweg neben dem mit sehr geringer Intensität auftretenden Molekülion als Basispeak das Ion 95, das unter Verlust zuerst der Methylgruppe am Kohlenstoffatom 1 und dann eines Propanonrestes gebildet wird (Dongmei et al., 2010). Als Nebenreaktion werden vom Molekül zuerst Wasser, dann eine Methylgruppe und anschließend eine Ethylengruppe abgespalten (Dongmei et al., 2010). Als Ausgangsverbindung wurde Bornylacetat gewählt. Die Oxidation zu 5-Oxobornylacetat erfolgte in einer Modifizierung der Methode von Acerson (2014). Abweichend von der dort beschriebenen Arbeitsweise wurde das Chromtrioxid wie bei Davydov et al. (2013) beschrieben als Feststoff zugegeben, da die bei Acerson (2014) beschriebene Konzentration des Oxidationsmittels in Essigsäure auf Grund der beschränkten Löslichkeit nicht hergestellt werden konnte. Die Zugabe erfolgte allerdings in kleineren Mengen und über einen längeren Zeitraum, da unter diesen Bedingungen die Ausbeute von 5-Oxobornylacetat steigt (Acerson, 2014). 5-Oxobornylacetat besitzt gegenüber dem von Davydov et al. (2013) hergestellten 5-Oxocampher den Vorteil von nur einer Oxogruppe und damit nur einer Angriffsstelle für die nachfolgende Reduktion und der damit verbundenen Einführung von Deuteriumatomen. Das Rohprodukt wurde nach destillativer Abtrennung von nicht umgesetzen Bornylacetat und anderen Nebenprodukten direkt der alkalischen Verseifung unterworfen, da 5-Oxoborneol auf Grund seines hohen Schmelzpunktes im Gegensatz zu 5-Oxobornylacetat sehr gut durch Umkristallisation gereinigt werden kann. Die Reduktion mit voll deuteriertem Hydrazinhydrat nach einer modifizierten Huang Minlon-Methode (Schwetlick, 2015) verläuft mit hoher Ausbeute. Bedingt durch die Keto-Enol-Tautomerie des Ausgangsproduktes 5-Oxoborneol ist vor der eigentlichen Reaktion ein Austausch der beiden Wasserstoffatome der zur Oxogruppe benachbarten Methylengruppe durch Deuteriumatome notwendig, da anderenfalls in Bezug auf die Deuteriumverteilung uneinheitliche Produkte erhalten werden. Die Oxidation von Borneol-d4 zu Kampher-d4 mit Hypochloriger Säure verläuft ebenfalls glatt und mit hoher Ausbeute. Die Synthese von 2-Methylisoborneol-d7 durch Reaktion von Kampher-d4 mit Trideuteromethylmagnesiumiodid liefert ohne Katalysator nur etwa 35 % 2-Methylisoborneol-d7 (Snoeyink et al., 1977), während der Großteil des Kamphers durch Enolatbildung nach der Aufarbeitung des Reaktionsgemisches unverändert verbleibt. Durch den Zusatz von Lanthanchlorid-Lithiumchlorid Katalysator (Knochel et al., 2009) konnte die Ausbeute auf etwa 85 % gesteigert werden. Leider entsteht dabei als Nebenprodukt eine unbekannte Substanz, die sich auch säulenchromatographisch nur sehr schwer und mit hohen Verlusten des Hauptproduktes abtrennen lässt.
Aus diesem Grund wurde die Aufreinigung bei einem Endgehalt von ca. 94 % an 2-MIB-d7 beendet. Die Gesamtausbeute an 2-MIB-d7 beträgt bezogen auf die Ausgangsverbindung Bornylacetat ca. 10 %, wobei die höchsten Verluste bei der Oxidation von Bornylacetat mit Chrom(VI)oxid zum 5-Oxobornylacetat und dessen Aufreinigung auftreten, während alle anderen Syntheseschritte mit mehr als 95%iger Ausbeute stattfinden. Das Massenspektrum entsprechend den obigen Ausführungen zeigt neben dem mit geringer Intensität auftretenden Molekülion und dem demethylierten Molekülion als intensivstes Ion die Masse 95. Außerdem werden auf Grund der Nebenreaktion die Ionen mit der Massenzahl 150, 135 und 107, allerdings ebenfalls mit relativ geringer Intensität gebildet (Abb. 5). Bei dem deuterierten Produkt beträgt der Massenshift des Hauptweges beim Molekülion 7, und bei den anderen beiden Ionen 4. Beim Nebenweg geht kein Deuterium verloren, so dass der Massenshift von 7 bei allen 3 Ionen der Fragmentierungsreaktion erhalten bleibt (Abb. 6).
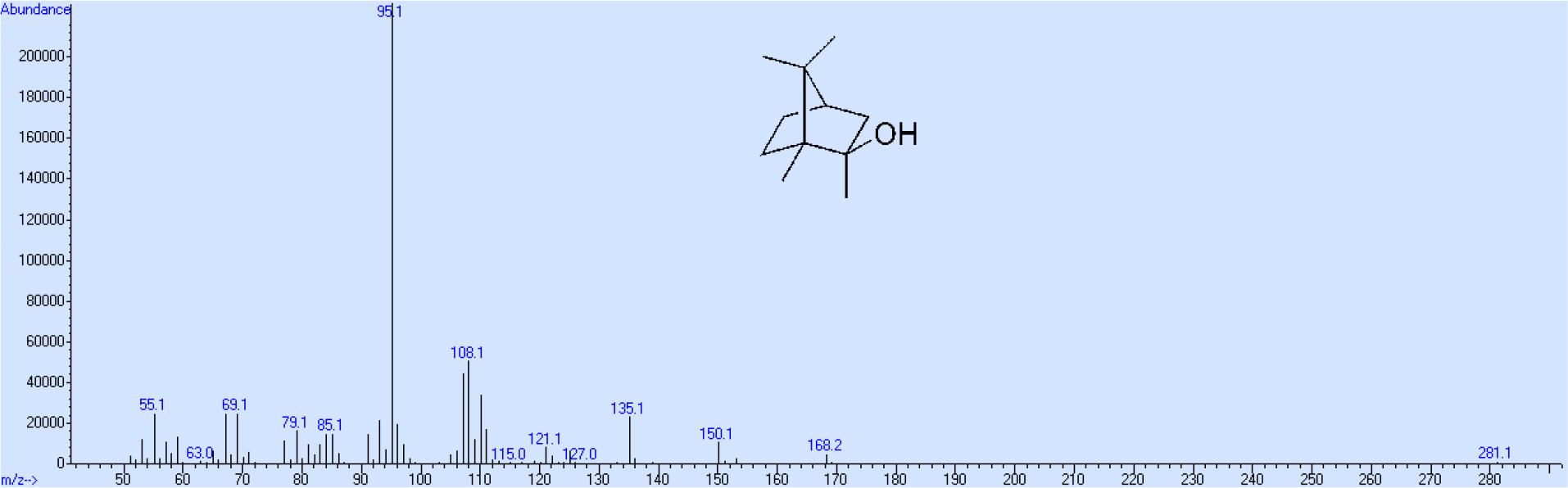
Massenspektrum von 2-Methylisoborneol
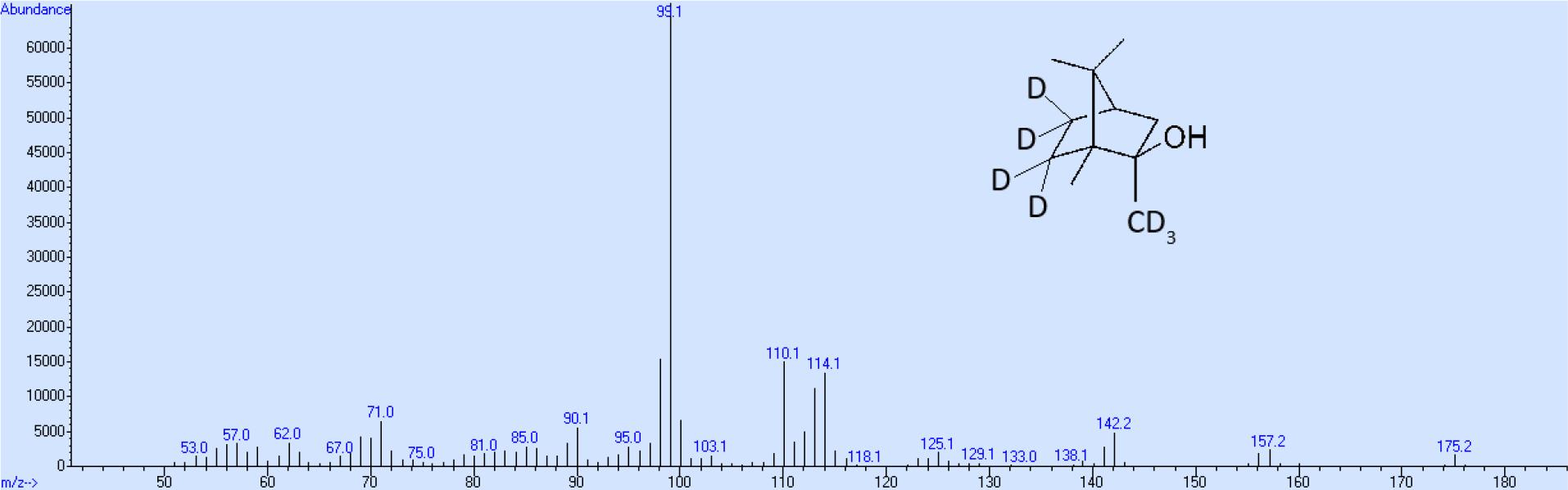
Massenspektrum von 2-Methylisoborneol-d7
Im Rahmen dieser Arbeit wurden die Verbindungen 2-Aminoacetphenon, 2,4,6-Tribromanisol und 2-Methylisoborneol in volloder teildeuterierter Form als interne Standards für die Quantifizierung der drei Verbindungen mittels HS-SPME hergestellt. Die Zielstellen für die Deuterierung wurden unter den Aspekten Vermeidung von Interferenzen zwischen internem Standard mit der Zielsubstanz, ausreichende Stabilität der Deuterierung unter den üblichen Probenvorbereitungs- und Messbedingungen sowie möglichst hohe Empfindlichkeit der Detektion ausgewählt. Die Synthesen von 2-Aminoacetophenon-d4 und 2,4,6-Tribromanisol-d5 sind durch leichte Zugänglichkeit der Ausgangsprodukte relativ problemlos zu bewerkstelligen. Hier ist lediglich bei den einzelnen Syntheseschritten auf eine Vermeidung des Verlustes von Deuterium zu achten. Die Synthese von 2-Methylisoborneol gestaltet sich dagegen wesentlich schwieriger. So ist das Ausgangsprodukt 5-Oxobornylacetat nur mit vergleichsweise geringer Ausbeute und unter Anwendung des sehr giftigen und carcinogenen Chrom(VI)oxids herstellbar. Für die Reduktion und gleichzeitiger Einführung der Deuteriumatome von 5-Oxoborneol zu Borneol erwies sich trotz vielfältiger Versuche lediglich die Huang-Minlon Variante als erfolgreich.
Zuletzt konnte eine nicht näher bekannte Verunreinigung des Rohproduktes von 2-Methylisoborneol mittels Säulenchromatographie trotz Verwendung verschiedener Laufmittelgemische nicht vollständig und nur unter hohen Verlusten am Zielprodukt abgetrennt werden. Der Vorteil von 2-MIB-d7 im Vergleich zu dem käuflichen 2-MIB-d3 besteht zum einen in der besseren gaschromatographischen Trennung von deuterierter und nicht deuterierter Substanz. So werden beispielsweise auch sehr hohe Konzentrationen von je 1μg/l von 2-MIB-d7 und 2-MIB auf einer unpolaren Säule basisliniengetrennt (Abb. 7). Außerdem ist bei der Verwendung von 2-MIB-d7 als interner Standard die mit Abstand intensivste Masse 95 für die Quantifizierung von 2-MIB auf Grund des Massenshiftes von 4 im Vergleich zur käuflichen Version von 2-MIB-d3, in der lediglich die Methylgruppe des C2-Atoms deuteriert ist, weil hier bei der Masse 95 kein Massenshift auftritt.
Leider gestaltete sich die Synthese von 2-MIB-d7 als unerwartet schwierig und zeitraubend, so dass keine Zeit für die entsprechenden Untersuchungen bezüglich Verbesserung der SPME-Analytik der Weinfehler blieb. Diese sollen in weiteren Arbeiten erfolgen.
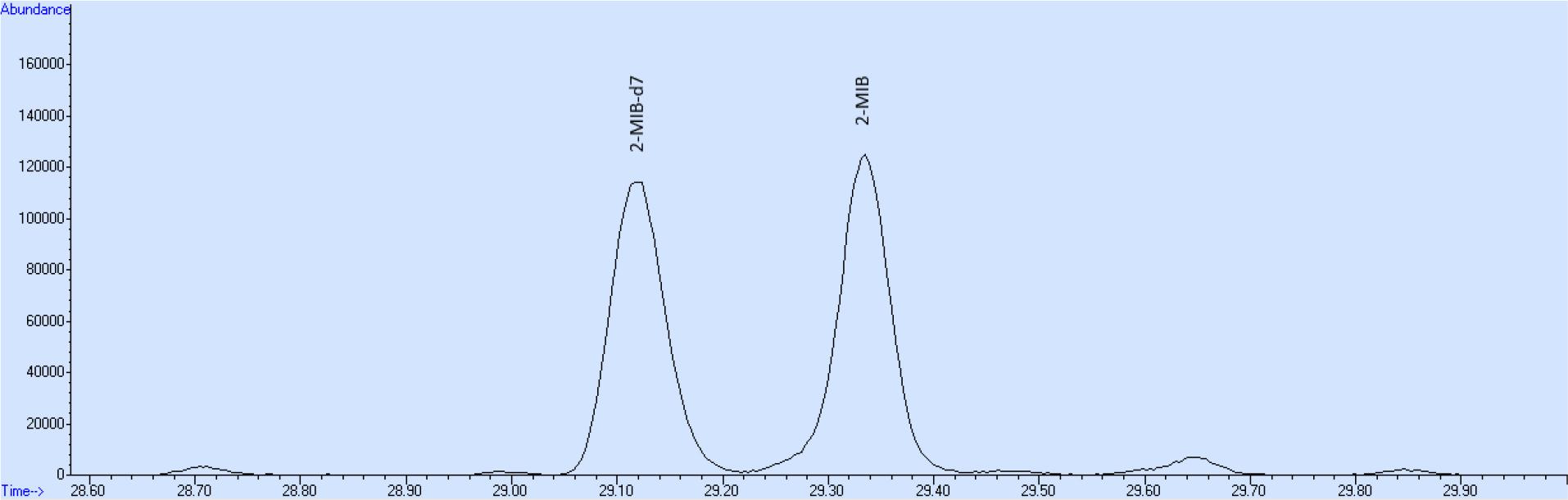
Chromatogramm einer wässrigen Lösung von 2-Methylisoborneol und 2-Methylisoborneol-d7 mit jeweils 1p.g/l Konzentration (Trennsäule DB-5, 60m, 0,25mmID, 0,25μmFD) mittels HS-SPME