
The diseases, some of which are parasitic, transmitted by arthropods such as ticks or fleas pose a serious health threat to pets and farm animals. Antiparasitic drugs are one of the largest groups of veterinary medications (9). Antiparasitic compounds include the isoxazoline derivatives that have been developed in the last 25 years. These include fluralaner, sarolaner, lotilaner, afoxolaner and esafoxolaner. A single oral dose of isoxazoline derivatives provides effective protection against arthropods for up to 12 weeks. These drugs have a rapid onset of action, which reduces the risk of disease transmission by ectoparasites (18, 19). In recent years, single and combined isoxazoline products have been approved by the American Food and Drug Administration and the European Medicines Agency, and these drugs are widely used to treat ectoparasite infections. Fluralaner, sarolaner and lotilaner have been approved for use in dogs and cats, afoxolaner has been approved for dogs (19), esafoxolaner for cats (15), fluralaner for hens (7, 11, 14) and lotilaner for humans (13). Despite the growing popularity of isoxazoline derivatives, very few studies have investigated their presence in biological matrices using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) (1, 2, 5, 10, 16, 17). Most of the described analytical methods support the determination of only one isoxazoline derivative in a given biological matrix (2, 5, 10, 16). These methods are also characterised by low repeatability due to incomplete information about equipment settings and extraction techniques. Therefore, the first aim of this study was to develop a sensitive, simple, inexpensive and rapid method for analysing isoxazoline compounds. The second aim was to revalidate the proposed method for use in human plasma and the plasma of three animal species, as well as in the first screening test of plasma obtained from a small animal clinic. Ultra-high performance (UP) LC-MS/MS was the technique selected to achieve the aims.
Blood samples for the analyses were obtained from 20 randomly selected, clinically healthy dogs (7 German shepherds and 2 golden retrievers aged 2–10 years; and 3 Yorkshire terriers, 3 schnauzers, 2 Siberian huskies, 2 beagles and 1 Labrador retriever, all aged 2–5 years) and from 10 randomly selected, clinically healthy cats (5 European and 5 Persian, all aged 3–10 years). Blood was not sampled specifically for the study, but was drawn during routine veterinary examinations with the owners’ consent. Blood that was not used for routine health examinations was donated to the researchers by courtesy of the owners and veterinary physicians. This method of acquiring biological material for research did not require the approval of the Ethics Committee. Blood was drawn from the brachial vein into heparinised sterile tubes (BD Biosciences, San Jose, CA, USA). Plasma was separated by centrifugation at 1,650 × g for 10 min at 4°C and was stored at -70°C until analysis.
Analyte-free plasma
obtained from clinically healthy animals and humans was thawed at room temperature and shaken in a vortex mixer at 1,000 rpm for 5 s. Plasma samples of 100 μL each were transferred to clean 5 mL polyethylene tubes and combined with 10 μL of fluralaner, sarolaner, lotilaner or afoxolaner and 10 μL of the fluralaner or sarolaner internal standard solution. The samples were shaken at 1,000 rpm for 5 s. Next, 450 μL of acetonitrile (ACN) : NH3 (96 : 4; v/v) was added for protein precipitation, and the samples were shaken at 3,000 rpm for 30 s. After centrifugation at 20,000 × g for 10 min at 4°C, 150 μL of the supernatant was passed through a 0.22 μm nylon syringe filter with a diameter of 13 mm (Waters, Milford, MA, USA) into chromatographic total recovery vials and injected into the LC-MS/MS system.
The reagents for LC-MS/MS, including ACN, NH3, methanol, water and formic acid (all LC/MS grade), were purchased from Sigma-Aldrich (Darmstadt, Germany). Analytical standards of fluralaner, sarolaner, lotilaner and afoxolaner (purity ≥98%) were supplied by HPC Standards GmbH (Borsdorf, Germany). Nitrogen for the LC-MS/MS system was supplied by a NitroGen N110R nitrogen generator (Peak Scientific, Inchanian, UK) and argon was supplied by Eurogaz-Bombi (Olsztyn, Poland).
The analyses were conducted using an Acquity UPLC system coupled to a XEVO TQ-XS triple quadrupole tandem mass spectrometer (Waters). Chromatographic separation was performed on an Acquity HSS (high-strength silica) T3 column (1.8 μm, 2.1 × 100 mm; Waters), and the mobile phase consisted of phase A (0.1% formic acid in ACN) and phase B (0.1% formic acid in water). The gradient elution programme of the mobile phase was as follows: 35% A in 0–2.6 min, 28% A in 2.7–2.9 min, 0% A in 3.0–3.4 min and 35% A from 3.50 min (Table 1). Each analysis lasted 5.5 min at a flow rate of 0.35 mL/min, including 3.5 min for chromatographic separation and 2 min for column re-equilibration. The injection volume was 1 μL, autosampler temperature was maintained at 15°C, and column temperature was maintained at 35°C. The MS/MS system was operated in electrospray positive ionisation mode (Table 1).
Selected parameters of ultra-high performance liquid chromatography–tandem mass spectrometry (MS/MS) for the determination of isoxazoline derivatives in plasma
| MS/MS parameters | Compound | ||||
|---|---|---|---|---|---|
| Fluralaner | Sarolaner | Lotilaner | Afoxolaner | ||
| Precursor ions (m/z) | 556.06 | 581.03 | 595.98 | 625.73 | |
| Product ions (m/z) | Quantification ions | 457.0 | 457.0 | 439.75 | 469.89 |
| Confirmation ions | 400.0 | 415.9 | 166.03 | 195.97 | |
| Collision energy (eV) | Quantification ions | 18 | 25 | 30 | 26 |
| Confirmation ions | 12 | 20 | 35 | 48 | |
| Retention time (min) | 2.55 | 2.55 | 3.33 | 3.18 | |
| Capillary voltage (kV) | 3.0 | ||||
| Cone voltage (V) | 20 | ||||
| Desolvation gas | Nitrogen | ||||
| Desolvation gas temperature (°C) | 600 | ||||
| Desolvation gas flow (L/h) | 1000 | ||||
| Collision gas flow (mL/min) | 0.2 | ||||
| Collision gas | Argon | ||||
| Source temperature (°C) | 120 | ||||
| Electrospray mode | Positive | ||||
| Dwell (s) | 0.1 | ||||
| Time (min) | Flow rate (mL/min) | Mobile phase (%) | Curve | ||
|---|---|---|---|---|---|
| A | B | ||||
| 0.00 | 0.35 | 35.0 | 65.0 | Initial | |
| 2.70 | 0.35 | 28.0 | 72.0 | 6 | |
| 3.00 | 0.35 | 0.0 | 100.0 | 6 | |
| 3.50 | 0.35 | 35.0 | 65.0 | 11 | |
| Mobile phase | 0.1% formic acid in acetonitrile | 0.1% formic acid in water | |||
| Column | ACQUITY HSS T3, 1.8 μm 2.1 × 100 mm | ||||
Stock solutions of fluralaner, sarolaner, lotilaner and afoxolaner were prepared by dissolving 0.5 mg of analytical standards in 5 mL of methanol. Sarolaner was the internal standard for fluralaner, lotilaner and afoxolaner, whereas fluralaner was the internal standard for sarolaner. The stock solutions were prepared in 5 mL volumetric borosilicate flasks (Duran Group, Mainz, Germany). A series of working standard solutions (all for the calibration curve) were prepared for validation by diluting the stock solutions with methanol. These solutions were prepared in 5 mL volumetric borosilicate flasks by diluting stock solutions in methanol at the following concentrations: 0.025, 0.125, 0.25, 0.625, 1.25, 2.5, 6.25, 12.5, 18.75 and 37.5 μg/mL. All solutions were stored at 4°C in the refrigerator and were brought to room temperature before use. Five calibration points solutions were additionally used to prepare quality control (QC) points: high QC (HQC) – 37.5 μg/mL, medium QC (MQC) – 25 μg/mL, intermediate QC (IQC) – 2.5 μg/mL (50% of the calibration curve), low QC (LQC) – 0.125 μg/mL and the lower limit of quantification (LLOQ) – 0.025 μg/mL.
The analytical method was validated as described by Kruve et al. (3, 4). The following parameters were determined during the validation procedure: linearity, accuracy, precision (repeatability/intra-day precision and intermediate precision/inter-day precision), limit of detection (LOD), LLOQ, selectivity, recovery, matrix effect, carry-over and stability (freeze– thaw stability, autosampler stability, working standard, stock stability and stability of sample processing temperature) (Table 2). All validation procedures were conducted in plasma sampled from clinically healthy laying hens. The method was then revalidated for selectivity, the matrix effect and total recovery based on the procedure described by Markowska et al. (6).
Ten calibration points were developed for the linearity test (0.001, 0.005, 0.01, 0.025, 0.05, 0.1, 0.25, 0.5, 0.75 and 1.5 μg/mL) for fluralaner, sarolaner, lotilaner and afoxolaner. The calibration points were prepared five times at daily intervals. Each curve was analysed twice, and each analysis was preceded by an analysis of a sample with no analytes (blank sample) and a sample containing only the internal standard (zero sample). The concentrations of isoxazoline derivatives in the plasma of laying hens were subjected to a linear regression analysis using the following regression equation: y = ax + b (Table 2).
Calculation methods and acceptance criteria for parameters for validation of an ultra-high performance liquid chromatography–tandem mass spectrometry method for the determination of isoxazoline derivatives in plasma
| Parameter | Acceptance criteria | |
|---|---|---|
| Linearity | Calibration points | Back-calculated concentrations should be within ± 15% of the nominal concentration, and at least 75% of the calibration points, but no fewer than 6, must fulfil this criterion |
| Coefficient of determination (r2) | ≥ 0.99 | |
| Relative residuals (Yi) | ||
| SD of relative residuals (SYi) | ||
| Stability | Stock and working standard | |
| Autosampler | ||
| Freeze and thaw | ||
| Sample processing temperature | ||
| Precision (RSD or CV) | ||
| Accuracy (deviation) (for at least 5 points per group/day) | ||
| LOD | 3 × SDCfortified where S/N ≥ 3: 1 | |
| LLOQ with accuracy and precision | 6 × Cfortified where S/N ≥ 10: 1 | |
| Matrix effect | ||
| Total recovery | ||
| Selectivity/Specificity | No endogenous peaks in the retention time of the analyte | |
| Carry-over | Area of carry-over peaks: ≤ 20% of LLOQ and 5% of IS area | |
yi – experimental signal; ŷi – calculated signal; SD – standard deviation; Yi – relative residual; Ȳ – mean value of relative residuals; St – peak area when the analysis was paused for time t; S0 – initial peak area without additional pauses during the analysis (freshly prepared standards); RSD – relative standard deviation; CV – coefficient of variation; Cmean – mean concentration (ng/mL); Ct – individually calculated concentration (ng/mL); Cn – nominal concentration (ng/mL); LOD – limit of detection; SDCfortified – standard deviation calculated from fortified samples with the lowest acceptable concentration; S/N – signal-to-noise; LLOQ – lower limit of quantification; Cfortified – minimal fortified concentration which meets the requirements; Xi – peak area of the analyte added to the matrix after extraction; X – peak area of the analyte in the final solvent; Xz – peak area of the analyte added to the matrix before extraction
Precision and accuracy were determined during daily analyses and between analytical dates. The QC points were prepared six times over a period of three days. Five calibration points were used to prepare QC as follows: HQC 37.5 μg/mL, MQC 25 μg/mL, IQC 1.5 μg/mL (50% of the calibration curve), LQC 0.125 μg/mL and LLOQ 0.025 μg/mL. It was specified for precision and accuracy for all QC points that they should not exceed ±15% of the nominal concentration and ±20% of the LLOQ (Table 2).
The stability of the studied isoxazoline compounds was estimated at all stages of sample storage, preparation and analysis. Stability tests were conducted after 3 h of storage at room temperature; after 24 h of storage in an autosampler at 4°C; after 24, 48 and 72 h of freezing/thawing at –80°C; and after 20 d of storage. Stability tests were prepared in six replicates for each QC point. Each QC sample was analysed relative to a freshly prepared calibration curve. The mean concentrations were within ±15% of the nominal concentration (Table 2).
The selectivity of the method was validated based on the presence or absence of interferences and by comparing the chromatograms of six blank plasma samples from laying hens, dogs, cats and humans, and blank serum samples spiked with the standard. The absence of interfering components was accepted when the response was 20% or less of the LLOQ for fluralaner, sarolaner, lotilaner and afoxolaner, and 5% or less for the internal standard (Table 2, Fig 1.).
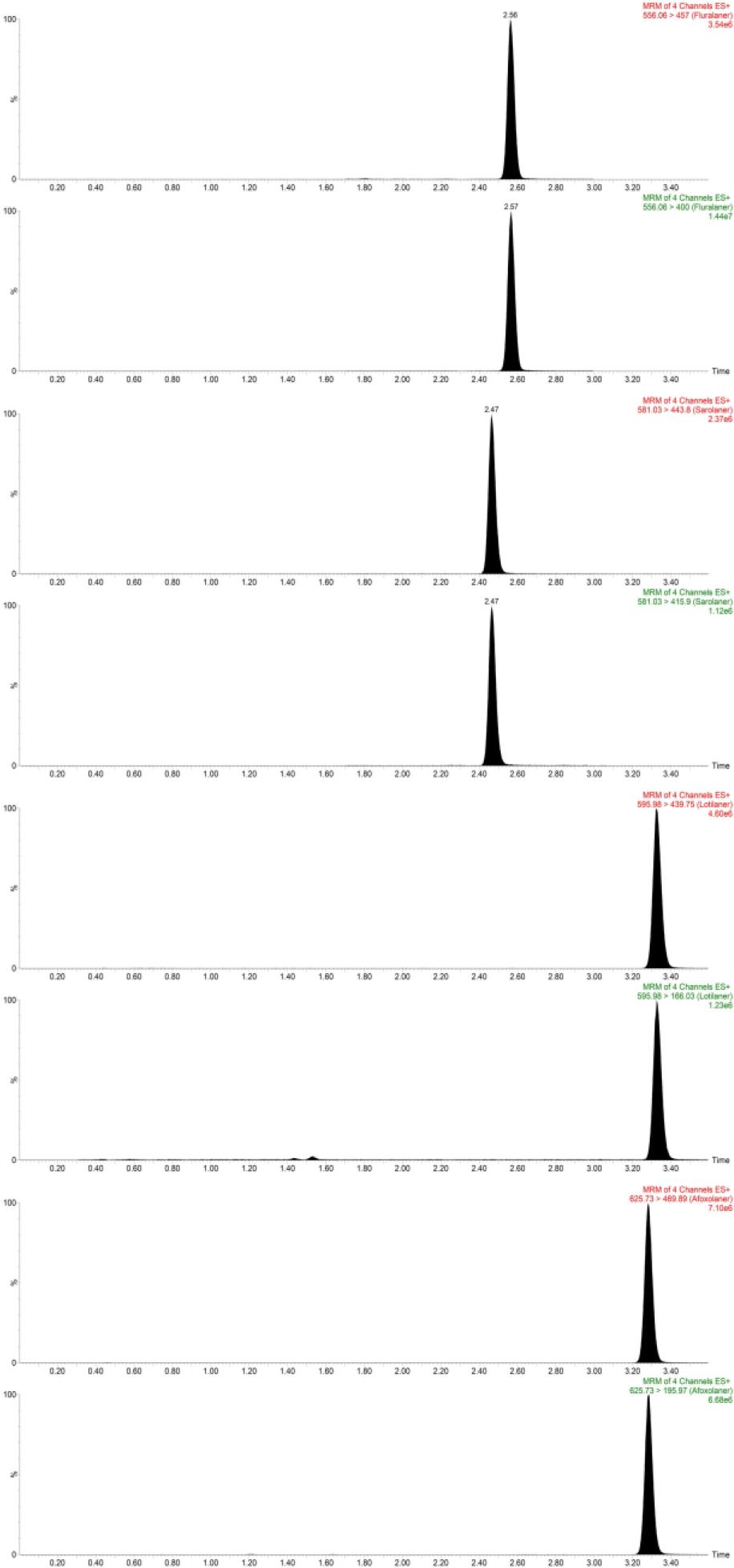
Liquid chromatography–tandem mass spectrometry traces of separation of four isoxazoline derivatives from plasma (fluralaner, sarolaner, lotilaner and afoxolaner)
Extraction recovery was assessed in six replicates of each QC point, where the analytical standards were added before and after extraction. The peak area of the analyte and the internal standard added after the extraction was indicative of 100% recovery (Table 2).
To evaluate the matrix effect, the analytical standards and the internal standard were added to plasma samples after extraction. Next, the signal from each compound was compared to the signal produced by fluralaner, sarolaner, lotilaner, afoxolaner and the internal standard that was added to a mixture of water (replacing plasma) and ACN : NH3 and was also prepared in six replicates for each QC point. It was specified that the increase or decrease in the extraction rate should not exceed ±15% (Table 2).
This test was conducted to eliminate any carry-over of isoxazoline derivatives between the samples in any part of the chromatography system (injector, column, mobile phase, etc.). The carry-over effect was assessed in six replicates by injecting a single blank sample after a HQC sample, and it was regarded as acceptable at ≤20% of the LLOQ (Table 2).
The mass spectrometer was operated in electrospray positive ionization (ESI+) mode. The spectra were traced in multiple-reaction-monitoring mode at 556.06 m/z to 400.0 and 457.0 for fluralaner, 581.03 m/z to 457.0 and 415.9 for sarolaner, 595.98 m/z to 439.75 and 166.03 for lotilaner and 625.73 to 469.89 and 195.97 for afoxolaner (Table 1).
The method developed in this study was fully validated in laying hen plasma. Additionally a validation outcome emerged from the results for total recovery and the matrix effect from samples of canine, feline and human plasma; these results are shown in Table 3. The results of the validation protocol met all acceptance criteria (6) (Table 2). The values of r and r2 in the linear regression analysis of the calibration curve were above 0.99 (Tables 4–7), and the entire calibration range was 0.001–1.5 μg/mL. Accuracy and precision were estimated for all QCs and the LLOQ, both for individual samples and for the six samples prepared over a period of three days. Intra-day precision was 1.79% to 14.97%, and accuracy was 1.33% to 11.67% for all QC and LLOQ (Tables 4–7).
Validation results for total recovery by ultra-high performance liquid chromatography–tandem mass spectrometry for the determination of isoxazoline derivatives in plasma from clinically healthy dogs, cats and humans and validation results for matrix effect
| Validation test | Dog | Cat | Human | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fluralaner | Sarolaner | Lotilaner | Afoxolaner | Fluralaner | Sarolaner | Lotilaner | Afoxolaner | Fluralaner | Sarolaner | Lotilaner | Afoxolaner | ||
| Total recovery (%) | Mean | 13.10 | 13.90 | 10.10 | 13.90 | 11.15 | 14.10 | 9.80 | 13.6 | 12.10 | 13.00 | 11.93 | 10.70 |
| SD | 2.10 | 1.80 | 1.18 | 2.00 | 1.43 | 1.12 | 2.50 | 2.20 | 2.90 | 0.80 | 0.90 | 1.10 | |
| Matrix effect (%) | Mean | 8.90 | 13.90 | 13.30 | 12.10 | 12.20 | 9.80 | 9.50 | 11.90 | 13.60 | 13.70 | 8.93 | 9.60 |
| SD | 1.54 | 1.89 | 1.04 | 2.03 | 2.12 | 2.34 | 2.12 | 1.07 | 1.14 | 2.01 | 1.08 | 1.71 | |
SD – standard deviation
Validation results of an ultra-high performance liquid chromatography–tandem mass spectrometry method for the determination of fluralaner in clinically healthy laying hen plasma
| Linearity | r2 | I 0.99 | II 0.99 | III 0.99 | IV 0.99 | V 0.99 | Mean 0.99 | |
|---|---|---|---|---|---|---|---|---|
| Precision (%) and accuracy (%) | Intra-day, n = 6; 3 replications | LLOQ | LQC | IQC | MQC | HQC | ||
| Precision | I | 8.94 | 5.59 | 1.94 | 3.89 | 1.66 | ||
| Accuracy | 6.67 | 4.33 | 1.62 | 2.64 | 1.29 | |||
| Precision | II | 4.15 | 6.81 | 2.34 | 7.81 | 2.47 | ||
| Accuracy | 1.67 | 5.33 | 1.90 | 4.65 | 1,99 | |||
| Inter-day, n = 18 | Precision | III | 7.66 | 9.38 | 5.54 | 3.25 | 6.16 | |
| Accuracy | 5.00 | 7.33 | 4.60 | 6.01 | 4,87 | |||
| Precision | 7.23 | 2.19 | 1.75 | 3.03 | 3.16 | |||
| Accuracy | 5.70 | 1.67 | 4.91 | 2.90 | 2.60 | |||
| Concentration (ng/mL) | S/N | |||||||
| LLOQ and LOD | LLOQ overall mean, n = 18 | 1.00 | 90.00 | |||||
| LLOQ overall SD, n = 18 | 0.14 | 5.91 | ||||||
| LOD overall mean, n = 18 | 0.5 | 6.22 | ||||||
| LOD overall SD, n = 18 | 0.33 | 3.91 | ||||||
| Total recovery | Sample | Total recovery (%) | ||||||
| Mean | 1 ng/mL | 91.19 | ||||||
| 1,500 ng/mL | 85.39 | |||||||
| SD | 1 ng/mL | 12.73 | ||||||
| 1,500 ng/mL | 5.94 | |||||||
| Matrix effect (%) | ||||||||
| Matrix effect | Mean | 1 ng/mL | 10.15 | |||||
| 1,500 ng/mL | 14.81 | |||||||
| SD | 1 ng/mL | 8.22 | ||||||
| 1,500 ng/mL | 2.94 | |||||||
| Stability tests results | ||||||||
| Stability | Period (h) | Changes in the concentration of QCs (%) | ||||||
| LQC | IQC | MQC | HQC | |||||
| 24 | 13.25 | 3.07 | 4.96 | 1.16 | ||||
| Working standard, 2°C | 48 | 8.92 | 14.58 | 12.01 | 14.28 | |||
| 120 | 13.25 | 10.90 | 9.02 | 12.24 | ||||
| 168 | 14.31 | 13.95 | 11.15 | 11.10 | ||||
| Autosampler, 4°C | 24 | 7.01 | 9.80 | 14.48 | 7.72 | |||
| 24 | 14.71 | 13.91 | 5.30 | 2.77 | ||||
| Freeze and thaw, –75°C | 48 | 14.07 | 14.93 | 12.60 | 4.65 | |||
| 72 | 12.33 | 14.39 | 13.98 | 13.49 | ||||
| 480 | 14.49 | 14.77 | 10.30 | 8.52 | ||||
| Sample processing, 21°C | 3 | 14.04 | 9.23 | 4.01 | 3.73 | |||
I–V – replicate number; LLOQ – lower limit of quantification; LQC – low quality control; IQC – intermediate quality control; MQC – medium quality control; HQC – high quality control; LOD – limit of detection; SD – standard deviation
Validation results of an ultra-high performance liquid chromatography–tandem mass spectrometry method for the determination of sarolaner in clinically healthy laying hen plasma
| Linearity | r2 | I | II | III | IV | V | Mean | |
|---|---|---|---|---|---|---|---|---|
| 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | |||
| Precision (%) and accuracy (%) | Intra-day, n = 6; 3 replications | LLOQ | LQC | IQC | MQC | HQC | ||
| Precision | I | 9.67 | 6.07 | 1.67 | 2.34 | 2,04 | ||
| Accuracy | 5.00 | 4.67 | 1.35 | 3.86 | 1,36 | |||
| Precision | II | 14.48 | 3.87 | 3.95 | 4.22 | 7,49 | ||
| Accuracy | 11.67 | 3.00 | 2.67 | 4.66 | 5,16 | |||
| Inter-day, n = 18 | Precision | III | 10.95 | 1.79 | 2.08 | 6.71 | 5,10 | |
| Accuracy | 6.67 | 1.33 | 1.65 | 7.08 | 3,33 | |||
| Precision | 11.70 | 3.91 | 2.57 | 6.02 | 4.88 | |||
| Accuracy | 7.78 | 3.00 | 1.89 | 3.33 | 3.28 | |||
| Concentration (ng/mL) | S/N | |||||||
| LLOQ and LOD | LLOQ overall mean, n = 18 | 1.00 | 91.48 | |||||
| LLOQ overall SD, n = 18 | 0.12 | 4.74 | ||||||
| LOD overall mean, n = 18 | 0.5 | 4.01 | ||||||
| LOD overall SD, n = 18 | 0.36 | 0.02 | ||||||
| Sample | Total recovery (%) | |||||||
| Total recovery | Mean | 1 ng/mL | 99.78 | |||||
| 1,500 ng/mL | 96.21 | |||||||
| SD | 1 ng/mL | 14.09 | ||||||
| 1,500 ng/mL | 7.47 | |||||||
| Matrix effect (%) | ||||||||
| Matrix effect | Mean | 1 ng/mL | 14.40 | |||||
| 1,500 ng/mL | 6.97 | |||||||
| SD | 1 ng/mL | 8.09 | ||||||
| 1,500 ng/mL | 7.00 | |||||||
| Stability test results | ||||||||
| Stability | Period (h) | Changes in the concentration of QCs (%) | ||||||
| LQC | IQC | MQC | HQC | |||||
| Working standard, 2°C | 24 | 13.97 | 12.24 | 12.35 | 13.31 | |||
| 48 | 12.08 | 14.38 | 13.55 | 14.44 | ||||
| 120 | 7.60 | 2.85 | 11.64 | 3.20 | ||||
| Autosampler, 4°C | 168 | 5.02 | 8.81 | 14.90 | 12.42 | |||
| 24 | 13.51 | 13.93 | 2.30 | 6.93 | ||||
| 24 | 7.12 | 14.11 | 14.92 | 7.17 | ||||
| Freeze and thaw, –75°C | 48 | 9.58 | 12.84 | 4.52 | 3.91 | |||
| 72 | 12.56 | 13.00 | 12.40 | 14.90 | ||||
| 480 | 7.16 | 2.30 | 2.66 | 10.25 | ||||
| Sample processing, 21°C | 3 | 13.38 | 14.62 | 3.21 | 4.07 | |||
I–V – replicate number; LLOQ – lower limit of quantification; LQC – low quality control; IQC – intermediate quality control; MQC – medium quality control; HQC – high quality control; LOD – limit of detection; SD – standard deviation
Validation results of an ultra-high performance liquid chromatography–tandem mass spectrometry method for the determination of lotilaner in clinically healthy laying hen plasma
| Linearity | r2 | I | II | III | IV | V | Mean | |
|---|---|---|---|---|---|---|---|---|
| 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | |||
| Precision (%) and accuracy (%) | Intra-day, n = 6; 3 replications | LLOQ | LQC | IQC | MQC | HQC | ||
| Precision | I | 14.97 | 1.79 | 3.67 | 4.46 | 7.82 | ||
| Accuracy | 11.67 | 1.33 | 2.58 | 6.72 | 5.60 | |||
| Precision | II | 7.40 | 4.10 | 4.95 | 5.02 | 4.61 | ||
| Accuracy | 5.00 | 3.29 | 3.27 | 7.03 | 3.76 | |||
| Inter-day, n = 18 | Precision | III | 6.32 | 2.33 | 5.99 | 8.22 | 1.42 | |
| Accuracy | 3.33 | 1.67 | 4.03 | 9.11 | 1.19 | |||
| Precision | 9.57 | 2.74 | 4.87 | 5.15 | 4.61 | |||
| Accuracy | 6.67 | 2.10 | 3.29 | 4.99 | 3.52 | |||
| Concentration (ng/mL) | S/N | |||||||
| LLOQ and LOD | LLOQ overall mean, n = 18 | 1.00 | 73.23 | |||||
| LLOQ overall SD, n = 18 | 0.09 | 5.95 | ||||||
| LOD overall mean, n = 18 | 0.5 | 5.55 | ||||||
| LOD overall SD, n = 18 | 0.39 | 2.87 | ||||||
| Sample | Total recovery (%) | |||||||
| Total recovery | Mean | 1 ng/mL | 95.50 | |||||
| 1,500 ng/mL | 93.16 | |||||||
| SD | 1 ng/mL | 14.76 | ||||||
| 1,500 ng/mL | 5.07 | |||||||
| Matrix effect (%) | ||||||||
| Matrix effect | Mean | 1 ng/mL | 14.10 | |||||
| 1,500 ng/mL | 7.09 | |||||||
| SD | 1 ng/mL | 5.98 | ||||||
| 1,500 ng/mL | 4.51 | |||||||
| Stability test results | ||||||||
| Stability | Period (h) | Changes in the concentration of QCs (%) | ||||||
| LQC | IQC | MQC | HQC | |||||
| Working standard, 2°C | 24 | 6.32 | 7.40 | 10.90 | 5.91 | |||
| 48 | 8.24 | 13.25 | 6.07 | 8.71 | ||||
| 120 | 9.58 | 13.60 | 2.96 | 11.92 | ||||
| Autosampler, 4°C | 168 | 8.45 | 14.87 | 11.29 | 12.82 | |||
| 24 | 13.50 | 13.99 | 9.54 | 14.94 | ||||
| 24 | 12.30 | 7.27 | 12.88 | 9.35 | ||||
| Freeze and thaw, –75°C | 48 | 8.75 | 5.60 | 11.63 | 12.44 | |||
| 72 | 13.03 | 2.37 | 3.95 | 6.73 | ||||
| 480 | 13.34 | 11.42 | 11.26 | 9.70 | ||||
| Sample processing, 21°C | 3 | 4.21 | 5.42 | 10.18 | 2.61 | |||
I–V – replicate number; LLOQ – lower limit of quantification; LQC – low quality control; IQC – intermediate quality control; MQC – medium quality control; HQC – high quality control; LOD – limit of detection; SD – standard deviation
Validation results of an ultra-high performance liquid chromatography–tandem mass spectrometry method for the determination of afoxolaner in clinically healthy laying hen plasma
| Linearity | r2 | I | II | III | IV | V | Mean | |
|---|---|---|---|---|---|---|---|---|
| 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | |||
| Precision (%) and accuracy (%) | Intra-day, n = 6; 3 repetitions | LLOQ | LQC | IQC | MQC | HQC | ||
| Precision | I | 7.66 | 10.28 | 3.04 | 2.47 | 3.45 | ||
| Accuracy | 5.00 | 8.67 | 2.48 | 3.22 | 2.00 | |||
| Precision | II | 13.22 | 4.20 | 4.30 | 6.66 | 8.16 | ||
| Accuracy | 10.00 | 2.67 | 3.27 | 5.54 | 6.39 | |||
| Inter-day, n = 18 | Precision | III | 9.67 | 3.10 | 2.26 | 5.23 | 4.52 | |
| Accuracy | 5.00 | 2.67 | 1.60 | 8.02 | 3.65 | |||
| Precision | 10.18 | 5.86 | 3.20 | 6.07 | 5.05 | |||
| Accuracy | 6.67 | 4.67 | 2.45 | 6.01 | 4.01 | |||
| Concentration (ng/mL) | S/N | |||||||
| LLOQ and LOD | LLOQ overall mean, n = 18 | 1.00 | 92.30 | |||||
| LLOQ overall SD, n = 18 | 0.04 | 4.8 | ||||||
| LOD overall mean, n = 18 | 0.5 | 0.9 | ||||||
| LOD overall SD, n = 18 | 0.44 | 1.11 | ||||||
| Sample | Total recovery (%) | |||||||
| Total recovery | Mean | 1 ng/mL | 85.19 | |||||
| 1,500 ng/mL | 87.05 | |||||||
| SD | 1 ng/mL | 14.14 | ||||||
| 1,500 ng/mL | 7.88 | |||||||
| Matrix effect (%) | ||||||||
| Matrix effect | Mean | 1 ng/mL | 13.77 | |||||
| 1,500 ng/mL | 14.69 | |||||||
| SD | 1 ng/mL | 9.56 | ||||||
| 1,500 ng/mL | 10.52 | |||||||
| Stability test results | ||||||||
| Stability | Period (h) | Changes in the concentration of QCs (%) | ||||||
| LQC | IQC | MQC | HQC | |||||
| Working standard, 2°C | 24 | 5.74 | 3.07 | 4.96 | 1.16 | |||
| 48 | 14.91 | 14.58 | 12.01 | 14.28 | ||||
| 120 | 9.82 | 7.82 | 11.76 | 14.80 | ||||
| Autosampler, 4°C | 168 | 12.79 | 3.31 | 11.28 | 13.84 | |||
| 24 | 13.15 | 8.10 | 9.55 | 14.14 | ||||
| 24 | 10.00 | 12.99 | 6.47 | 3.44 | ||||
| Freeze and thaw, –75°C | 48 | 14.10 | 13.78 | 14.05 | 12.39 | |||
| 72 | 14.84 | 3.12 | 13.48 | 10.89 | ||||
| 480 | 12.53 | 10.90 | 12.66 | 8.37 | ||||
| Sample processing, 21°C | 3 | 5.66 | 14.37 | 13.26 | 13.68 | |||
I–V – replicate number; LLOQ – lower limit of quantification; LQC – low quality control; IQC – intermediate quality control; MQC – medium quality control; HQC – high quality control; LOD – limit of detection; SD – standard deviation
Inter-day precision was 1.75% to 11.70%, and accuracy was 1.67% to 7.78% for all QC and LLOQ (Tables 4–7). The LOD was set at 0.5 ng/mL ± 0.38 (with a minimum signal-to-noise ratio of 10 : 1) and the LLOQ was 1.0 ng/mL ± 0.04–0.14 (Tables 4–7). The analysis of the selectivity/specificity of the method revealed no significant peaks during the retention of isoxazoline derivatives in drug-free plasma separated from the blood of clinically healthy animals. No significant carry-over of fluralaner, sarolaner, lotilaner or afoxolaner was observed in analyses involving high concentrations of these analytes. All analytes appeared to be stable after 3 h of storage at sample processing temperature; after 24 h of storage in an autosampler at 4°C; after 24, 48, 72 and 480 h of thawing and freezing cycles; and after 168 h of refrigerated storage at 2°C (Tables 4–7). The mean matrix effect for all species was 11.99%.
The proposed method was successfully applied to analyse the plasma of randomly selected patients of a small animal clinic for the presence of isoxazoline derivatives. The concentrations of isoxazoline compounds were within the analytical range of the method in 15 out of 20 dogs (1.0–434.6 ng/mL) and in 2 out of 10 cats (23.8–233.3 ng/mL) (Table 8).
Results of screening tests (concentrations in ng/mL)
| Species | Breed | Fluralaner | Sarolaner | Lotilaner | Afoxolaner |
|---|---|---|---|---|---|
| Dog | German shepherd | - | 434.6 | - | - |
| German shepherd | - | 12.1 | - | - | |
| German shepherd | - | - | - | - | |
| German shepherd | 414.3 | - | - | - | |
| German shepherd | - | - | - | 1.3 | |
| German Shepherd | - | 19.0 | - | - | |
| German shepherd | 346.5 | - | - | - | |
| Yorkshire terrier | - | - | - | 1.0 | |
| Yorkshire terrier | - | - | - | 1.0 | |
| Yorkshire terrier | - | - | - | 1.0 | |
| Schnauzer | 388.3 | - | - | - | |
| Schnauzer | 76.7 | - | - | - | |
| Schnauzer | - | - | - | - | |
| Golden retriever | - | - | - | - | |
| Golden retriever | - | 225.0 | - | - | |
| Siberian husky | - | - | - | - | |
| Siberian husky | - | 196.0 | - | - | |
| Beagle | - | - | - | - | |
| Beagle | - | 5.6 | - | - | |
| Labrador retriever | - | 100.6 | - | - | |
| Cat | European cat | - | - | - | - |
| European cat | - | 23.8 | - | - | |
| European cat | - | 233.3 | - | - | |
| European cat | - | - | - | - | |
| European cat | - | - | - | - | |
| Persian cat | - | - | - | - | |
| Persian cat | - | - | - | - | |
| Persian cat | - | - | - | - | |
| Persian cat | - | - | - | - | |
| Persian cat | - | - | - | - |
The present study was undertaken to develop the first ever method for determining four isoxazoline compounds in plasma samples. The proposed method was developed in several steps. The optimal detector parameters were selected in the first step; the optimal chromatographic conditions were selected in the second step; and a quick, effective, easy and inexpensive analyte extraction method was developed in the third step. In the fourth step, the proposed method was validated in laying hen plasma and revalidated in canine, feline and human plasma. The validated method was successfully used in a screening study to determine the concentrations of isoxazoline derivatives in dogs and cats that were randomly selected in a private veterinary clinic.
The molecular weight of isoxazoline derivatives was 556.29 g/mol for fluralaner, 581.36 g/mol for sarolaner, 596.76 g/mol for lotilaner and 625.87 g/mol for afoxolaner. These values were used to determine the parent ions of the analysed molecules on the assumption that each substance was ionised only once (m/z = 1/1). The optimal chromatographic conditions were selected in the following stage of the study. First, the column was selected based on the properties of isoxazoline derivatives which are lipophilic compounds (XLogP3 of 5.6 for fluralaner, 3.4 for sarolaner, 6.6 for lotilaner and 6.7 for afoxolaner). Shoop et al. (15) used a Zorbax SB C18 column (2.1 mm × 50 mm, 5 μm) to determine the plasma concentrations of afoxolaner, whereas Toutain et al. (16) determined the plasma levels of lotilaner using a Daicel Chiralpak IA-3 column (150 × 4.6 mm) with a much larger particle size than that applied in the current study. A Zorbax Eclipse Plus C18 column (4.6 × 100 mm, 3.5 μm) was used only by Wu et al. (17) to measure the concentrations of all isoxazoline compounds, but in their study, tissues rather than plasma were used as the matrix. In contrast, the column used in the present study had a smaller particle size to achieve higher analytical sensitivity. The Acquity HSS T3 column was also chosen because it is compatible with 100% aqueous solvents. A similar mobile phase to that used in the present experiment was applied by Sari et al. (8), but they used methanol instead of acetonitrile. Methanol was not used in this study because it leads to a maximal reduction of baseline resolution. Wu et al. (17) used a mobile phase composed of 5 mmol/L of aqueous ammonium formate solution and MeOH. In the present study, however, a buffer was not used to avoid crystallisation, which may occur under mass spectrometry conditions due to solvent evaporation and elevated temperatures in the ion source.
Sample preparation is a critical step in analyses of drug residues. An adequate sample preparation protocol removes matrix interference and improves the sensitivity and robustness of the method. The main goal of this study was to develop a quick, easy, short, sensitive, and low-cost method of plasma purification. Protein precipitation is a short procedure that was well suited to the needs of this study. To date, a short and easy method for extracting lotilaner from plasma has only been proposed by Toutain et al. (16). In turn, Kilp et al. (2) and Sari et al. (8) used pure ACN for protein precipitation and determined the plasma levels of fluralaner by solid-phase extraction. Wu et al. (17) also used ACN for extraction, followed by a cleanup step involving a PRiME (protein removal in mass spectrometry experiments) HLB (hydrophilic–lipophilic balance) cartridge, and they analysed four isoxazoline compounds (similarly to the present study), but in tissues, not in plasma, which explains the additional cleanup step. For this reason, the method developed by the cited authors is longer, consists of more steps and is more expensive to validate when adopted for tissue analysis because of the high cost of tissue samples.
The method was successfully validated according to Kruve et al. (3, 4). The calibration curves showed linearity in a sixfold wider range than other methods (1, 8, 17). The validation confirmed sensitivity several times higher than that described by other authors (1, 17). The validated method demonstrated acceptable inter-day precision and accuracy across all QC levels and LLOQ. In contrast, Elliot et al. (1) reported lower precision, with inter-day values exceeding 19.5% and falling out of the ranges of all parameters except LLOQ in our protocol. The developed method demonstrated high analyte stability under all tested conditions, whereas Elliot et al. (1) and Wu et al. (17) did not include stability testing, for example during freeze–thaw cycles, which may limit the long-term reliability of their quantification results. In contrast to Wu et al. (17), Sari et al. (8) and Elliot et al. (1), we evaluated the selectivity of our method. This avoided any potential interferences and carry-over effects during analysis, especially because our calibration curve included very high and very low concentration points. The mean matrix effect for all species was not quite 12%, confirming that the developed method was influenced by matrix components to an acceptable extent by the criteria established.
The developed analytical method met all validation criteria, demonstrating superior sensitivity, precision and stability compared with data available in the literature. Minimal matrix effect, low LOD/LLOQ and comprehensive stability tests indicated greater reliability of this method for accurately measuring isoxazoline derivatives. The proposed method was successfully used to analyse the plasma of randomly selected cats and dogs from a small animal clinic for the presence of isoxazoline derivatives. This screening confirmed that the method worked well under real clinical conditions, and could be taken up for the detection and measurement of isoxazoline residues in plasma collected during routine veterinary visits. Such a screening test would reveal whether antiparasitic drugs had been used and give a veterinarian reliable information when there was no record of treatment. Therefore, the presented method can be useful not only in pharmacokinetic studies, but also for control purposes, especially in cases of suspected drug side effects or misuse, or of their undocumented administration. Given the growing use of isoxazoline-based treatments, developing a fast, cost-effective and reproducible screening method is essential for accurate monitoring and regulatory compliance.
Although the developed method met the criteria selected by the authors, the main limitation of the study was that the MS/MS protocol was designed to examine each isoxazoline compound separately. Another is that no isotopic internal standard was used in the study although such a standard would have been a better choice in tandem mass spectrometry to obtain more consistent results. In addition, the sample preparation method used in the study is a “dirty” technique that can shorten the life of the column. Despite these limitations, the proposed method is a reliable solution for determining isoxazoline derivatives in plasma. Further research is needed to address the identified shortcomings and achieve more satisfactory results.
This is the first study to propose and validate a UPLC-MS/MS method for quantifying all isoxazoline derivatives in the blood plasma of laying hens, dogs, cats and humans. The results indicate that the developed method is replicable, precise, accurate, selective and sensitive. The main advantages of the method are its simplicity and effectiveness, as well as quick sample preparation, which decreases costs and facilitates rapid and precise determination of the analytes in plasma. Despite the existence of cleaner techniques of plasma purification than protein precipitation, the proposed method had a high recovery rate, and the matrix effect was small enough for the method to be validated with sufficient accuracy and precision. The method seems to be suitable for practical use, and was successfully applied to test for the isoxazoline derivatives fluralaner,sarolaner, lotilaner and afoxolaner in animals that had been orally administered isoxazolines.