
Secretory breast carcinoma (SBC) is a rare and distinctive histological subtype of breast cancer, accounting for approximately 1% of all breast malignancies [1]. Despite its rarity in the general population, SBC represents nearly 80% of primary breast cancers in children and adolescents, making it the most frequent malignant breast tumor in this age group [1,2]. The entity was originally described in 1966 by McDivitt and Stewart under the name “juvenile breast carcinoma” [2]. Subsequent reports established that SBC can occur in both sexes and across all age groups, which led Tavassoli and Norris to propose the more accurate descriptive term “secretory carcinoma” [3].
Clinically, SBC typically presents as a slow-growing, painless, retroareolar mass, often detected incidentally during physical examination [1,4, 5, 6]. Pediatric cases have been described from as early as 3 years to late adolescence, with a mean age at diagnosis of around 9–12 years [6,7]. While the clinical course in children is usually indolent, the risk of local recurrence is well documented and may occur even decades after the initial treatment [7,8]. Although uncommon, distant metastases have been reported, particularly in adult patients, and are associated with a less favorable prognosis [9].
Histologically, SBC is defined by lobular growth with microcystic, tubular, and solid architectural patterns, as well as abundant eosinophilic secretory material within intracellular vacuoles and glandular lumina [3,10]. Immunohistochemically, SBC is characteristically positive for S-100, MUC4, and GATA3. Hormone receptor status is variable: although many tumors are reported as triple-negative, partial estrogen receptor (ER) positivity has been described in several pediatric and adult cases, suggesting phenotypic heterogeneity [5,8,11].
The most prominent molecular characteristic of SBC is the recurrent ETV6-NTRK3 fusion gene [4]. This trans-location leads to the expression of an in-frame chimeric protein with constitutive tyrosine kinase activity, resulting in aberrant activation of MAPK and PI3K/AKT signaling pathways that drive tumorigenesis [12]. Importantly, the detection of ETV6-NTRK3 not only provides diagnostic specificity but also opens therapeutic opportunities. Targeted TRK inhibitors such as larotrectinib and entrectinib, and more recently repotrectinib, have demonstrated high response rates in both adult and pediatric patients with NTRK-fusion positive tumors, including SBC [6,13,14].
Despite the generally favorable outcome, the rarity of the disease and lack of standardized treatment guidelines complicate management. Surgery remains the mainstay of therapy, but the optimal extent is debated: while breast-conserving surgery is desirable, mastectomy is often favored in children due to high recurrence risk and limited residual breast tissue [1,7,15]. Radiotherapy is usually avoided because of the potential for long-term toxicity, including secondary malignancies, while chemotherapy has not shown consistent benefit [9,16].
Here we present a six-year-old girl with secretory breast carcinoma, with complete clinicopathological and molecular characterization, including confirmation of the ETV6-NTRK3 fusion. By integrating histological, immunohistochemical, and genetic findings, this report contributes to the limited pediatric SBC literature and outlines the importance of multidisciplinary management and molecular profiling in rare pediatric tumors.
A six-year-and-three-month-old previously healthy girl was admitted to our center with a palpable retroareolar mass in the left breast, measuring approximately 10 × 10 mm. The lesion had first been noticed six months earlier and demonstrated a slow but steady increase in size. There was no history of trauma, pain, nipple discharge, or inversion. On examination, the mass was firm, mobile, and painless, with intact overlying skin. No axillary lymphadenopathy was detected, and the contralateral breast appeared normal. The child’s physical development was appropriate for age, and there was no personal or family history of malignancy. Routine hematological and biochemical investigations were un-remarkable. Preoperative ultrasonography was inconclusive, and the initial clinical impression was that of a fibroadenoma.
An open excisional biopsy was performed. Histopathological examination revealed a lobulated tumor composed of atypical epithelial cells with eosinophilic cytoplasm and large vesicular nuclei with prominent nucleoli. The tumor exhibited microcystic, cribriform, and tubular patterns, as well as solid nests of cells. While nuclear pleomorphism was mild, mitotic activity was prominent, with seven mitoses per ten high-power fields. Intracytoplasmic and luminal PAS- and d-PAS–positive secretory material was observed.
Immunohistochemical studies demonstrated strong positivity for S-100, MUC4, EGFR, and GATA3, with focal CD117 expression. The estrogen receptor was positive in ~20% of tumor cells, while the progesterone receptor, HER2, SMA, and p40 were negative. Ki-67 labeling index was approximately 20%, with moderate p53 nuclear staining (70%) and rare p63 positivity (5%). Histopathological findings are illustrated in Figure 1 (a–g).

Gross, histopathological, and immunohistochemical features of secretory breast carcinoma in a six-year-old girl: (a) Gross specimen of the excised tumor, approximately 10 × 10 mm, well-circumscribed and lobulated; (b) Lobular tumor architecture with a clearly visible pseudocapsule (HE, ×25); (c) Microcystic, cribriform, and tubular growth patterns with intraluminal secretory material (HE, ×100); (d) High mitotic activity, reflecting the proliferative potential of the tumor (HE, ×1000); (e) Focal membranous/cytoplasmic CD117 positivity (×200); (f) Ki-67 nuclear positivity in approximately 20% of tumor cells (×200); (g) Rare scattered p63-positive nuclei (<5% of tumor cells) (×200); (h) Strong and diffuse nuclear/cytoplasmic S-100 immunopositivity (×100).
Given the patient’s age, germline genetic testing was performed to exclude hereditary cancer predisposition. No pathogenic variants were identified in BRCA1 or BRCA2, nor in an extended multigene panel including ATM, BRIP1, CDH1, CHEK2, NBN, PALB2, PTEN, RAD51C, RAD51D, STK11, and TP53. Tumor molecular profiling, however, revealed the canonical ETV6-NTRK3 in-frame fusion (ETV6 exon 5 – NTRK3 exon 13), confirming the diagnosis of secretory breast carcinoma at the molecular level. This alteration is widely recognized as the oncogenic driver in SBC and represents a predictive biomarker for responsiveness to TRK inhibitors.
Because the excisional biopsy showed a positive margin, the patient underwent a simple mastectomy according to standard oncologic principles. All margins were negative, and the sentinel lymph node biopsy revealed no evidence of metastasis. The postoperative course was uneventful, and no adjuvant radiotherapy or chemotherapy was administered.
At six-month follow-up, ultrasonography revealed two enlarged axillary lymph nodes (22 × 18 mm and 6 × 2 mm) with heteroechoic structure and increased vascularization, raising suspicion of progression. Axillary lymphadenectomy was performed, but all nodes were histologically benign (Figure 2).

Intraoperative photograph during axillary lymph node dissection performed at six-month follow-up due to two ultrasonographically suspicious nodes (22 × 18 mm and 6 × 2 mm), which were subsequently confirmed to be histologically benign.
Six years after the initial surgery, the patient remains free of local recurrence or distant metastasis (Figure 3).
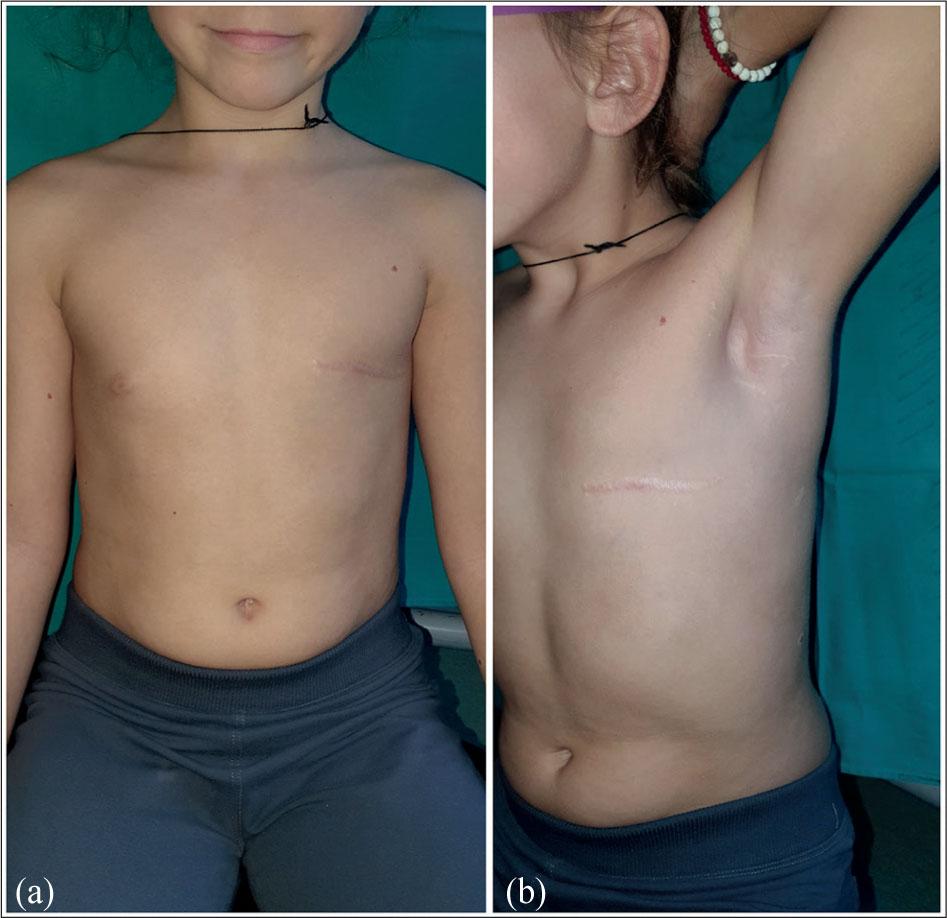
Long-term postoperative follow-up of the patient: (a) Frontal view demonstrating good cosmetic outcome after mastectomy; (b) Slightly angled frontal view, confirming a well-healed scar and symmetrical chest contour.
Importantly, the detection of ETV6-NTRK3 fusion provides a therapeutic safety net: in the event of relapse or systemic progression, targeted therapy with TRK inhibitors such as larotrectinib, entrectinib, or repotrectinib would represent a viable treatment option.
SBC remains rare, with fewer than 100 pediatric cases reported to date [1,16]. This rarity makes every new case valuable for refining diagnostic and therapeutic strategies. Histopathological and immunohistochemical features in our patient—lobular architecture, microcystic and cribri-form patterns, PAS-positive secretions, S-100 and MUC4 positivity, as well as partial ER expression—are consistent with those described in previous reports [3,8,9]. Interestingly, although SBC has historically been classified as a “triple-negative” tumor, our case again demonstrates that partial ER positivity can be encountered, underscoring the immunophenotypic heterogeneity of this entity. Such variability further emphasizes the importance of combining histology with molecular diagnostics to achieve diagnostic certainty.
Management of pediatric SBC remains controversial. While local excision is often appealing for preserving breast tissue, mastectomy is frequently favored in children due to the higher risk of recurrence and the relatively limited amount of breast tissue available for re-excision if margins are positive [1,7,13]. This was precisely the challenge in our patient: the initial excisional biopsy revealed positive margins, prompting a definitive mastectomy. Although this approach is more radical, it offered oncological safety in a prepubertal girl where preservation of breast development was not feasible. Radiotherapy is generally avoided in this age group due to the substantial risk of radiation-induced malignancies and long-term growth disturbances [17]. Likewise, adjuvant chemotherapy has not demonstrated consistent benefit in SBC [9,16], reflecting the indolent natural history of the disease and its relative chemoresistance.
The identification of the canonical ETV6-NTRK3 fusion in our patient is of particular importance. This genetic alteration not only confirms the diagnosis of SBC but also aligns the tumor with a broader biological spectrum that includes congenital fibrosarcoma and cellular mesoblastic nephroma, where the same fusion drives oncogenesis [4]. This molecular convergence supports the idea of SBC as part of a family of “ETV6-NTRK3 fusion-driven tumors,” characterized by aberrant activation of MAPK and PI3K/AKT pathways, leading to sustained proliferation and survival signals. Importantly, pediatric patients with NTRK-fusion–positive tumors have shown remarkable responses to TRK inhibitors, such as larotrectinib and entrectinib, with high response rates and durable remissions [6,7,18].
Although our patient is currently disease-free six years post-surgery, the presence of the ETV6-NTRK3 fusion provides a clear therapeutic avenue in the event of relapse or metastatic progression. The availability of histology-agnostic targeted therapies marks a paradigm shift: rather than relying on nonspecific cytotoxic regimens, treatment could be tailored based on the underlying oncogenic driver. In this sense, molecular profiling transforms SBC from a surgically managed rare entity into a candidate for precision oncology. Our case, therefore, illustrates how genetic insights not only enrich diagnostic accuracy but also empower clinicians and families with actionable options for the future.
This case highlights the importance of combining histopathology with molecular diagnostics in pediatric breast cancer. The identification of ETV6-NTRK3 fusion not only confirms the diagnosis but also provides a therapeutic pathway if relapse occurs. Surgery remains the cornerstone of therapy, but targeted treatments represent an evolving paradigm. Reporting such cases contributes to the limited global experience and aids in refining clinical guidelines.