
Few scientific questions are as profound as the origin of life. One of the biggest mysteries in origin of life research concerns the emergence of today’s biopolymers. Unraveling this question requires careful consideration of early Earth environments. Geological and geochemical evidence points toward highly dynamic settings, characterized by strong temperature gradients and fluctuating hydration states, which are continuously driving chemical systems out of equilibrium (1,2,3,4,5,6,7,8). Such non-equilibrium environments force molecular systems to explore broader chemical space, favoring reactions and pathways that would be hindered under constant, fully aqueous conditions (9,10,11).
Wet-dry cycling is one of the key non-equilibrium processes proposed to promote molecular complexation on early Earth (9, 12,13,14,15,16,17,18,19,20). In shallow aqueous environments, such as small ponds or surface pools, repeated evaporation and rehydration act as a simple yet powerful physical driver of chemical evolution (21). During drying phases, condensation-dehydration reactions are favored, whereas during rehydration, more labile products are selectively hydrolyzed. This constant and repeated pressure introduces a rudimentary form of selection, favoring molecular structures that can persist and accumulate over prolonged timescales.
Peptide bond formation has been demonstrated via several mechanisms under plausible prebiotic conditions (22,23,24). Despite their central role in modern biology, amino acids do not readily form peptide bonds in aqueous environments; peptide bond formation is thermodynamically disfavored by competition with hydrolysis and kinetically limited by the high activation energy required for amide bond formation. These limitations have led to the proposal of numerous pathways invoking solutions such as activated monomers, catalysts, or constrained environmental reaction conditions.
A conceptually elegant solution to peptide bond formation emerges by considering peptide formation in environments undergoing wet-dry cycling, proceeding through an exchange reaction rather than a direct condensation between two amino acid monomers. Modern biology itself relies on this strategy: ribosomal peptide synthesis proceeds through activated ester intermediates rather than direct amide bond formation (25). Examination of the ribosomal peptidyl transferase centre (PTC) reveals two functionally distinct sites: the P site and the A site (26,27,28,29,30). The P site holds the growing peptide chain esterified to a transfer RNA (tRNA), while the A site accommodates an incoming aminoacyl-tRNA poised for incorporation into the elongating peptide. Notably, peptide bond formation in this context does not proceed via a direct condensation-dehydration reaction. Instead, elongation occurs through an ester-amide exchange reaction, in which the amine of the amino acid in the A site nucleophilically attacks the ester linkage between the peptide and the tRNA in the P-site (31). These chemical parallels are consistent with the protoribosome hypothesis, which proposes that a primitive RNA-based catalytic scaffold was capable of promoting peptide bond formation before the advent of genetically encoded translation (26, 27, 31). Support for this view comes from structural evolutionary analyses of the ribosome (25, 32,33,34). These studies suggest that the ribosome evolved in an accretionary, ‘onion-like’ manner, in which new structural layers were added over time without substantial loss or rearrangement of the ancestral core. The peptidyl transferase center, located at the heart of the ribosome, appears to be among the most ancient components, with progressively younger structural and functional elements radiating outward. This layered architecture preserves a molecular record of evolutionary history, offering a plausible timeline by which a simple RNA-based catalytic scaffold could have gradually acquired increasing complexity while retaining its original chemical function.
Importantly, such a protoribosomal system would not require sequence specificity or templated information transfer, but only the spatial organization of activated substrates. Structural and mechanistic analyses of the modern ribosomal peptidyl transferase center suggest that it functions primarily as an entropic and geometric catalyst rather than a chemically active enzyme, relying on precise positioning of reactants to facilitate ester-amide exchange. This observation raises the possibility that a much simpler assembly, lacking coded specificity yet capable of binding activated substrates, could plausibly have supported early peptide elongation (and possibly elongation of other proto-polymers). In this view, the ribosome is not an abrupt evolutionary innovation but the refined descendant of a prebiotic chemical system that already exploited ester-mediated peptide formation.
A similar chemical trajectory to peptide bond formation in the ribosome can be recapitulated in reactions of mixtures of hydroxy acids and amino acids under wet-dry cycling conditions. During dehydration, hydroxy acids readily form esters via condensation-dehydration reactions (13, 35,36,37,38,39). In the presence of amino acids, subsequent ester-amide exchange reactions generate depsipeptides, which are heterogeneous polymers containing both ester and amide linkages (16, 17, 40,41,42,43,44,45,46,47,48). Notably, this process requires no sophisticated catalysts (49) and emerges naturally from the environmental cycling of prebiotically available building blocks (hydroxy acids and amino acids) (50,51,52,53,54).
Furthermore, within these fluctuating wet-dry environments, primitive RNA/pre-RNA oligomers or RNA-peptide assemblies capable of transient substrate binding could have further biased ester-amide exchange reactions toward peptide formation. Rather than merely driving peptide bond formation, wet-dry cycling functions as a chemical filter that selects for certain molecular traits. Across repeated wet-dry cycles, a gradual enrichment of peptide bonds relative to ester bonds has been observed, consistent with the greater resistance of amide linkages to hydrolytic stress compared with ester bonds (17). In this regime, the environment itself biases the system toward peptide-rich polymers. Hence, mere chemical stability, rather than biological function, emerges as an early dominant selective pressure shaping polymer composition.
While plausible routes to peptide formation have been explored, several key questions remain unresolved. Most prominent among these are - why biology converged on a limited subset of 20 α-amino acids, how amino acid homochirality might have emerged, and how early polymers gradually acquired increased structural and functional complexity (55,56,57,58,59,60,61,62). In this regard, several studies have highlighted depsipeptides as experimentally tractable models for early chemical evolution of peptides. For example, Cronin group has demonstrated that recursive wet-dry cycling of hydroxy acids and amino acids, resulting in depsipeptide formation, can give rise to emergent function. In that work, esterase activity arose only after repeated wet-dry cycles, underscoring the importance of environmental recursion and non-equilibrium conditions for functional emergence, in contrast to static reaction conditions. Moreover, work by Williams, Leman, Hud, and colleagues has shown the selective incorporation of proteinogenic over nonproteinogenic cationic amino acids into depsipeptides under drying conditions, providing evidence that modern biochemical building blocks could have been enriched through purely chemical selection mechanisms (44). As another example, a recent work from our group revealed a selection mechanism favoring α-backbone over β-backbone architectures in depsipeptide libraries, driven by their propensity to assemble into stable droplets via liquid-liquid phase separation (63). This finding suggests that basic physicochemical constraints may have biased early polymer composition. In addition, Hud and colleagues explored depsipeptide nucleic acids as potential prebiotic alternatives to RNA, demonstrating that ester-amide-based backbones can support base pairing and molecular recognition, thereby expanding the landscape of plausible early informational polymers (64).
Overall, wet-dry cycling provides an experimentally tractable and conceptually coherent framework for exploring the prebiotic formation of polymers such as peptides and peptide analogs. This platform has also been utilized for the formation of other molecules, such as polyesters (36, 37), nucleic acids (64), amino acid-keto acid conjugates (65), and oligosaccharides (66). Altogether, integrating insights from modern biology with prebiotic chemistry allows us to probe not only the origins of life, but also the chemical and physical constraints required for life to emerge (67,68,69). From this perspective, the emergence of the ribosome can be understood not as the origin of peptide synthesis, but as its refinement. Environmental wetdry cycling, ester-mediated exchange chemistry, and simple catalytic scaffolds together define a continuum in which early, non-coded peptide formation gradually transitioned toward the highly regulated translational machinery observed today (Figure 1). The protoribosome concept thus provides a conceptual link between prebiotic polymer chemistry and extant biology, reinforcing the idea that modern biochemical complexity arose through incremental stabilization and organization of pre-existing chemical processes.
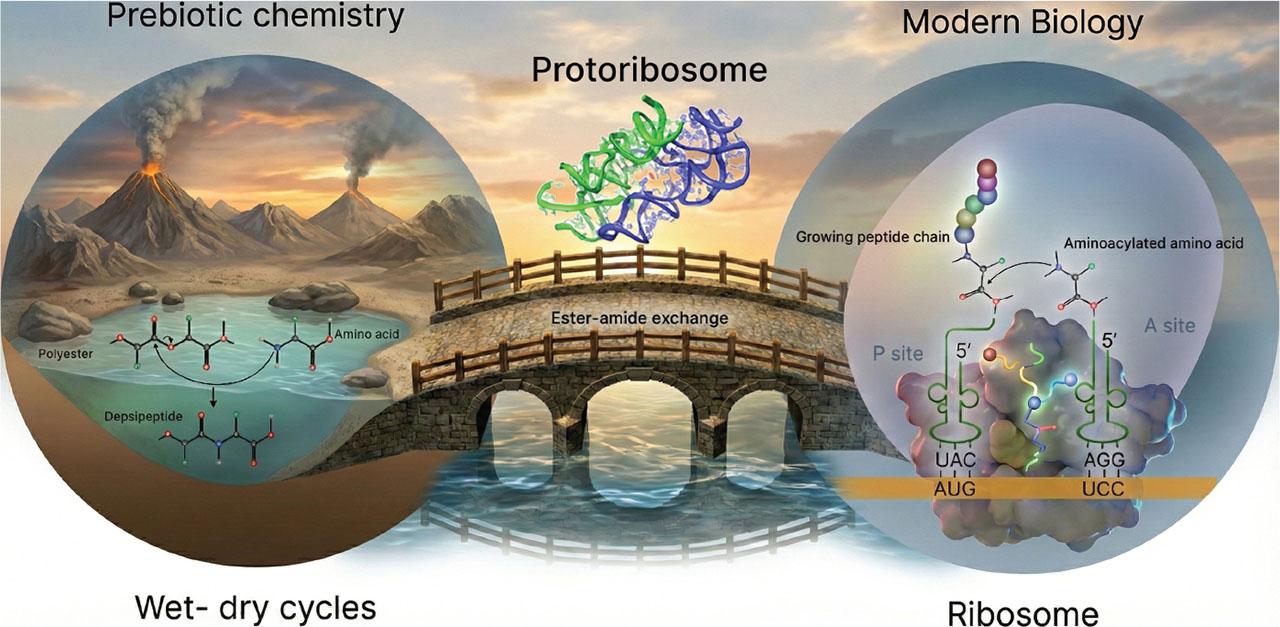
Conceptual link between prebiotic chemistry and modern biology. Ester–amide exchange reactions occurring in a small aqueous pond enable peptide formation, paralleled by an analogous ester–amide exchange reaction in the ribosome that elongates the peptide chain during protein synthesis – all connected by the chemical evolution of a protoribosome complex.
Although the precise historical sequence by which life emerged on Earth 4 billion years ago will remain inaccessible to direct reconstruction, contemporary studies outline chemically and physically plausible pathways consistent with early terrestrial conditions. In this context, the systematic interrogation of extant biochemistry serves not merely as a retrospective inquiry into our evolutionary origins, but as a means of identifying the underlying constraints, regularities, and organizing principles that render the emergence of living systems possible. Such work reframes the problem of life’s origin from a singular historical event to a question about the fundamental laws and boundary conditions under which matter can transition into self-sustaining, evolving complexity.