
It is estimated that 20% of the general population has asymptomatic hyperuricemia (aHU). According to the data from the National Health and Nutrition Examination Survey 2007–2016, the incidence of hyperuricemia (HU) remained stable in the United States (Chen-Xu et al. 2019). In 2022, allopurinol (ALL) had been prescribed more than 15 million times in the US alone. These data leave no doubt that uric acid lowering therapy (ULT) is one of the most prescribed therapies in developed countries.
HU is independently associated with the risk of chronic kidney disease (CKD), diabetes, metabolic syndrome, or hypertension (Chen et al. 2019; Wu et al. 2021; Maloberti et al. 2023). In rat models, HU is linked to hypertension, higher proteinuria and creatinine values, renal hypertrophy, and glomerulosclerosis (Kang et al. 2002). Logically, lowering serum uric acid (sUA) should improve cardiovascular prognosis and renal outcomes. As implementing dietary restrictions reduces sUA only by 0.5–2 mg/dL, ULT is the only way to achieve desired sUA levels. However, despite abundant data on the subject, the question of whether aHU should be treated remains an open one. Sato et al. (2019) published a review in Nature, proposing routine measurements of serum urate levels in patients with CKD and initiation of ULT in aHU with deteriorating renal function. This opinion was based on the pathophysiology of UA influence on renin-angiotensin-aldosterone system, intracellular oxidative stress or crystalline effects, and the heterogeneity of clinical trials. The authors deemed several trials noninterpretable because the control group did not show clinically relevant progression of CKD and argued that follow-up time over 12 months shifted the trial results toward improvement of kidney function. They have also underlined that available genetic studies focus on gene polymorphisms of urate transporters and not intracellular urate levels. A year later, Gonzalez-Martin et al. (2020) published an extensive analysis exposing increased all-cause mortality in patients with ULT and debating unreliable results of pro-ULT trials: high crossover between ULT and control groups, changed definition of a renal event in a post hoc analysis, lack of an appropriate control group, or statistical insignificance.
In our opinion, such differences between esteemed researchers stem from the heterogeneity of data: (a) lack of one, precise definition of HU; (b) influence of genetic and environmental factors on an individual’s sUA level; (c) possible use of other (better?) markers of HU than sUA; (d) lack of an adequate animal model to study HU; and (e) imperfect estimated glomerular filtration rate (eGFR) calculation. Heterogenous primary and secondary end points in clinical trials, inadequate follow-up, and a wide range of statistical methods used to analyze data are some of the variables that have an impact on our understanding of HU. Finally, the amount of data is overwhelming, and meta-analyses bring conflicting results.
We do not aim to answer the ultimate question of “treat or not to treat.” In our review, we analyzed the research on HU in CKD to give the reader an update on the current trends and the results of newly published studies. Initially, we showed the current perception of aHU, then we debated possible influence of HU on kidneys, and we ended by proposing the future directions for HU studies. For the clarity of the text, aHU is referred to as “HU,” by which we mean “higher than normal sUA level without any evidence of tissue damage.” We did not focus on uric acid (UA) related to acute kidney injury (AKI) or on cardiological indications for ULT, as these are a separate topic.
In short, HU is the concentration of UA molecules at which urate crystals start to aggregate in laboratory conditions. The definition is based on the solubility limit of urate in bodily fluids, which depends on urate concentration, temperature, pH, and the presence of the following nucleation centers: indicators (albumins, globulins – particularly gamma-globulin type 1, and collagen 1 in synovial fluid) and reductors of nucleation (ions K+, Cu2+, and Mg+). Urate remains in solution indefinitely at a concentration of 5 mmol/L (Merriman 2024). However, given different measuring techniques (automated enzymatic vs. colorimetric methods, providing results higher by approximately 1 mg/dL), the definition of HU varies from 6 mg/dL to >8 mg/dL (Mount 2023). Putative harmful effects of elevated sUA levels on renal disorders probably occur at concentrations that are subsaturating and do not cause gout.
Interestingly, large trials on HU had inclusion criteria ranging from >4.5 mg/dL to >7.1 mg/dL (Gonzalez-Martin et al. 2020), while some studies report values as high as 9.8 mg/dL as the best mortality predictors (Hare and Johnson 2003). The question arises whether the cutoff value for HU diagnosis should be personalized and dependent on (a) gender – as the increased mortality is associated with sUA >7 mg/dL in men and >5 mg/dL in women (Konta et al. 2020); (b) climate and ethnicity (Curhan 2021); (c) presence of comorbidities (Ulutaş et al. 2021); and (d) the underlying cause of HU. Without a clear cutoff value, even if it is specific for certain populations, no conclusions can be drawn regarding the treatment indications and goals.
HU stems from either increased purine biosynthesis or a decreased clearance. UA overproduction can be related to diet, cytotoxic drug use, lymphoproliferative disorders, hemolysis, psoriasis, down syndrome, or inherited enzyme defects. Low clearance of UA can be caused by CKD, the use of certain drugs (thiazides, loop diuretics, aspirin, calcineurin inhibitors, vitamin B3, or bempedoic acid), volume depletion, diabetic ketoacidosis, lactic acidosis, obesity, high circulating insulin, and monogenic disorders such as uromodulin (UMOD) mutation and variants of encoding transporter genes (Mount 2023). If the background is unclear or the patient does not respond adequately to ULT, a genetic cause can be suspected, and one should consider further investigation. Evaluation includes placing the patient into one of the following groups: (a) renal underexcretion type (RUE), (b) renal overload type (ROL), (c) combined type, or (d) normal type (Nakayama et al. 2020). Importantly, this is the only classification supported by GWAS studies (Matsuo et al. 2016b; Nakayama et al. 2020). Two parameters are needed to place the patient into one of the mentioned groups: (1) fractional excretion of UA (FEUA), which is the percentage of the urate filtered by glomeruli that is excreted in the urine, calculated by dividing the ratio of urine to plasma urate by the ratio of urine to plasma creatinine
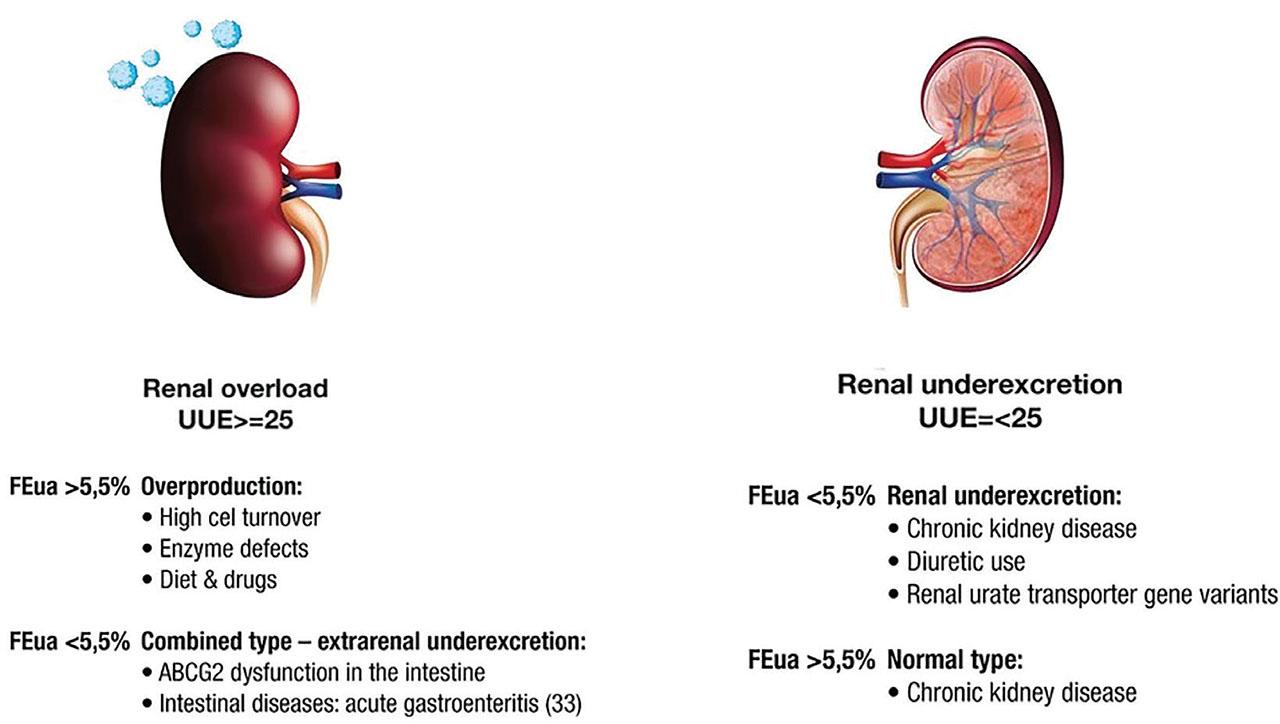
Types of HU. FEUA, fractional excretion of uric acid (unit: %), HU, hyperuricemia; UUE, urinary urate excretion (unit: mg/h/1.73 m2).
Contrary to popular belief, diet explains very little variation in sUA levels in the general population (Major et al. 2018). Renal underexcretion with low FEUA is considered the predominant type of HU (Perez-Ruiz et al. 2002). UA overexcretion is uncommon, and renal overload is responsible for only 10%–20% of HU (Curhan 2021). However, a recent study found that dysfunction of the ABCG2 gene appears as often as in 75.6% of the Japanese population. Additionally, heritability of fractional excretion of UA is estimated between 46% and 96%, which underlines the role of single nucleotide polymorphisms (SNPs) in the determination of patients’ UA levels (Narang et al. 2019). Very little can be found on the “normal type” as it is only briefly mentioned in the original (Nakayama et al. 2020) and review papers (Ichida et al. 2012; Dalbeth et al. 2019). Surprisingly, as Li et al. (2021) showed in their research, in patients with CKD, FEUA increases greatly, from 6.8% in CKD stage 3 to 19.5% in CKD stage 5. According to the authors, this result may reflect the compensation by residual nephrons as the rate of glomerular filtration determines the UA level in advanced CKD, as opposed to renal tubular reabsorption and secretion in healthy kidneys. This topic is hardly mentioned in other studies. Perhaps, the high variability of research results stems from the fact that sUA fails as the ultimate marker of tissue damaging HU. Strong evidence supports hyperuricosuria as a cause of kidney injury, but it is not clear whether HU without hyperuricosuria is also harmful (Gonzalez-Martin et al. 2020). Surrogate biomarkers of HU, such as FEUA, UUA, clearance of UA, glomerular filtration load of UA (Guan et al. 2020), and novel biomarkers, such as UMOD/creatinine ratio (Wu et al. 2019) or interleukin (IL)-37 (Ding et al. 2021) should be defined, validated, and measured prospectively during various interventions reducing CKD progression. Moreover, it came to our attention that only one large study measured GFR instead of estimating it with mathematical equations (Doria et al. 2020). This can farther contribute to bias in large-scale studies as eGFR calculated with the CKD-EPI equation is not validated for patients with certain conditions, for example, high body mass, very early or late CKD stages, diabetes, or kidney transplant. Measuring GFR should be the standard procedure when planning trials aimed at settling long-disputed subjects. Additionally, studies should not only rely on GFR to assess kidney function. Other biomarkers such as albumin-creatinine ratio, neutrophil gelatinase-associated lipocalin, kidney injury molecule 1, or transforming growth factor (TGF)-β1 can provide valuable information about kidney damage (Perrenoud et al. 2020).
There is a significant difference in the inflammatory process causing gout and hypothetical mechanism causing chronic kidney injury. Metabolic and renal effects of HU are mediated by intracellular urate (an ionized form of UA, dominating in the blood). A drop in pH shifts the balance toward UA in the form that later can cause gout. In normal pH of the blood, urate stimulates NADPH oxidase and migrates to the mitochondria. This increases the intracellular oxidative stress, even though extracellular UA acts as an antioxidant (reviewed by Sato et al. [2019]). In gout, monosodium-urate (MSU) crystal deposition triggers the formation of inflammasomes, consisting of three domains (NACHT, LRR, and PYD) containing protein 3 (NLRP3) and an adaptor protein. The inflammasome is activated by caspase 1, on which it starts to produce IL-1β and IL-18, further inducing the production of secondary inflammatory mediators (tumor necrosis factor [TNF]-α, IL-6, and IL-8) and recruitment of neutrophils. Neutrophils are central to the positive feedback loop of inflammation: crystal phagocytosis and degranulation, direct crystal lysis, activation of monocytes/macrophages and complement pathways, and release of mediators of pain and tissue damage (Dalbeth and Haskard 2005). Finally, the flare is resolved in part thanks to NETs, which are neutrophil extracellular trap structures, formed from neutrophils after they have undergone an oxidative burst. In an environment with high neutrophil densities, NETs capture and degrade cytokines and chemokines via serine proteases. Interestingly, NET form dense aggregates that resemble gouty tophi (Schauer et al. 2014). Novel medications (anti-IL-1 and IL1β, anakinra, and canakinumab) in gout management are aimed at the first part of the inflammatory process, but with the growing knowledge, we expect even more targeted approaches.
In aHU, no acute tophi formation is observed. Ponticelli et al. (2020) hypothesized that the abundant UA enters epithelial cells through one of the transporters and then damages the cells in two mechanisms (Figure 2). Inside the cell, it triggers oxidative stress by increasing renin-angiotensin-aldosterone and NADPH activity and reduces nitric oxide production. Reactive oxygen harms the endothelial cells and induces the proliferation of smooth muscle cells, resulting in hypoxia and then fibrosis. The second pathway is mediated by macrophages and dendritic cells. In receptor-mediated and independent ways, intracellular UA activates the inflammasome. Increased production of ILs, cytokines, and toll-like receptor mediates injury, resulting in fibrosis, vascular remodeling, vascular resistance, and increased sodium absorption (Ponticelli et al. 2020).
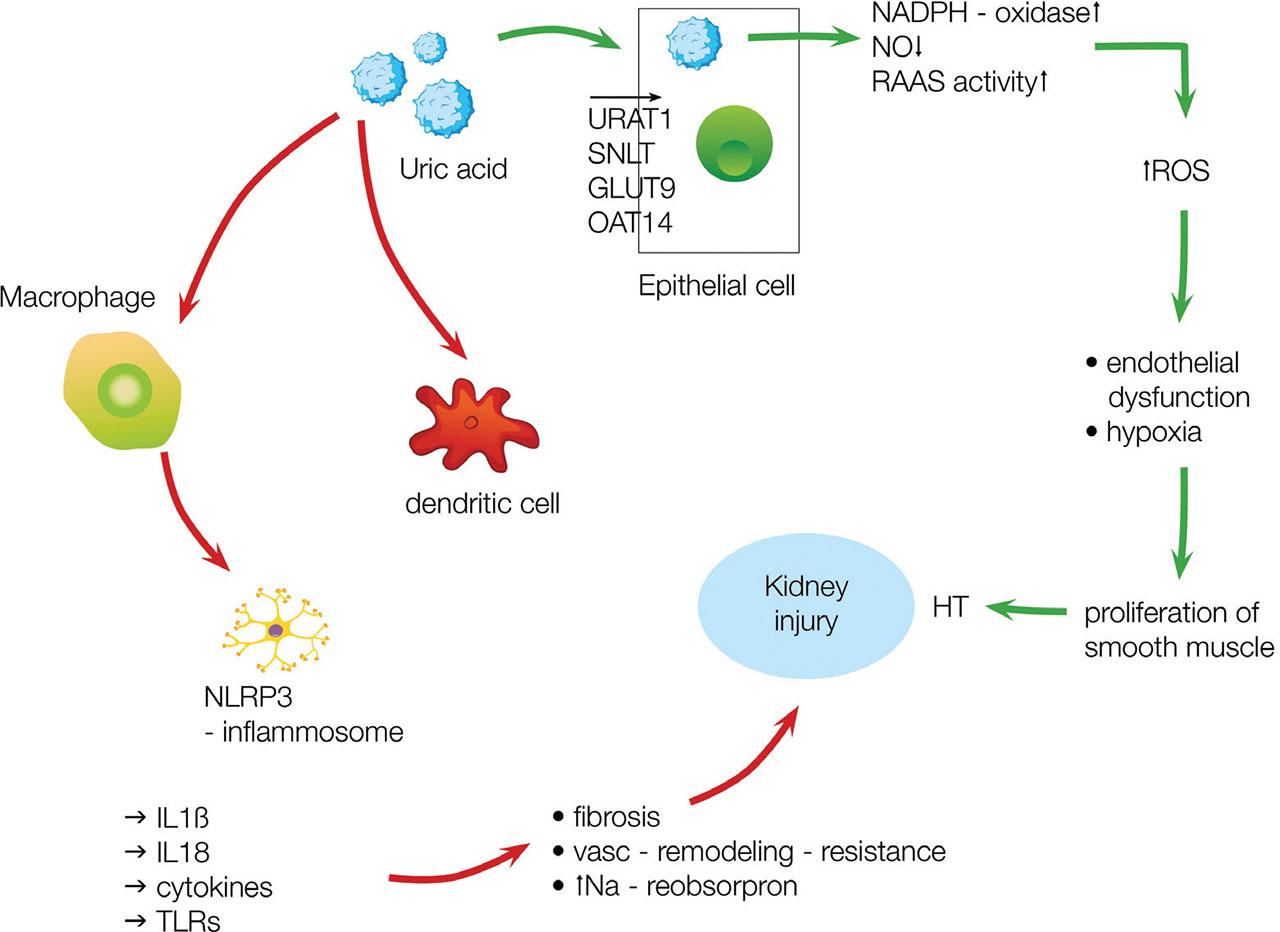
UA contributes to the kidney injury by nonimmunological (green arrows) and immunological (red arrows) pathways. Process description in the main text. IL, interleukin; UA, uric acid; URAT1, urate transporter gene 1. Adapted from Ponticelli et al. (2020).
A pathological effect of chronic exposure to increased sUA concentration has not been fully proven in humans. In cell lines and animal models, HU induces kidney injury both during acute or chronic exposure (Johnson et al. 2018). In rats, HU activates the NLRP3 inflammasome, synthesis of TGF-β1 and NF-κB signaling pathways, and the expression of multiple profibrogenic cytokines (Liu et al. 2015; Hu et al. 2022). Animals develop microvascular and inflammatory models of kidney injury, including afferent arteriolar disease, which impairs blood pressure regulation and reduces blood flow. This can lead to CKD and/or aggravation of a preexisting disease. However, animal-model findings cannot be simply extrapolated to human subjects. So far, the histopathological changes in animals have not been reproduced in human subjects, e.g., in afferent arteriole resistance (Lu et al. 2019; Gonzalez-Martin et al. 2020). Additionally, not all animal models have shown the destructive effects of HU on CKD. Sellmayr et al. (2020) used a mouse model with CKD induced by aristocholic acid. The assessment was performed using mass spectrometry, immunohistochemistry, confocal microscopy, and flow cytometry. The conclusion was that HU alone did not cause or exacerbate CKD. Only HU with UA crystalluria due to urinary acidification caused tubular obstruction, inflammation, and interstitial fibrosis (Sellmayr et al. 2020). This is also known for humans with genetic defects of tubular urate transport (SLC22A12) when hyperuricosuria may cause nephropathy even if blood results indicate hypouricemia.
Whether the deposition of urate crystals in the tubular lumen and interstitium in the outer medulla can cause CKD in humans has not been confirmed. Conversely, focal crystal deposition was in the past, considering an unlikely cause of diffused tubulointerstitial fibrosis, microvascular lesions, and glomerulosclerosis (Moe 2010; Piani and Johnson 2021). More recent studies named HU as a risk factor for segmental glomerulosclerosis and tubular atrophy/interstitial fibrosis, even when sensitivity and specificity models have adequate accuracy only for the first one (Fan et al. 2019). Gouty tophi in kidneys are difficult not only because crystals dissolve in formalin but also able because renal biopsies target the renal cortex, and even in medullary tissue, tophi are reported only in up to 4.5% of renal specimens (Ayoub et al. 2016), which could bias experimental research. In a widely commented study, Bardin et al. (2021) brought back the idea of microcrystalline nephropathy. Among 502 Vietnamese patients with gout, 36% showed a hyperechoic pattern of Malpighi pyramids on B-mode ultrasonography in comparison to the kidney cortex. No such pattern was observed in a matching control group without gout. Hyperechoic kidney medulla was frequent in patients with tophaceous gout and associated with features of tubulointerstitial nephritis (Bardin et al. 2021). Similar cases of superficially “asymptomatic” HU have been recently described in other tissues (Howard et al. 2011; Pineda et al. 2011; Dalbeth et al. 2015).
Frequently, the presence of kidney stones is associated with UA-related tissue damage and prompts clinicians to introduce a UA-lowering therapy. This is questionable, as UA stones account for only 5%–10% of urinary tract stones in the United States and Europe, while the risk of both UA and calcium oxalate stones is increased in patients with gout. UA stones are much more common (up to 40%) in hot and dry climate, which predispose to dehydration and acidic urine pH (Curhan 2021) or in patients suffering from chronic diarrhea with bicarbonate loss and volume depletion, for example, after colonic resection. The assessment of a patient with HU and nephrolithiasis should include stone analysis or, if not possible, the urinary pH and 24-h urine collection, before introducing ULT.
According to American College of Rheumatology, indications for ULT include the following: the presence of subcutaneous tophi, radiographic damage attributable to gout, gout flares more than once a year, and a gout flare in a patient with CKD stage ≥3, SU >9 mg/dL or urolithiasis. An additional indication for ULT is the prevention of tumor lysis syndrome. According to the Kidney Disease: Improving Global Outcomes guidelines, aHU does not require treatment, though this statement is marked as “low evidence.” Allergic reaction to urate-lowering drugs (ULDs) is the main contraindication. ALL, mostly associated with cutaneous adverse reactions, is strongly contraindicated in patients with known HLA-B*5801 allele (the society recommends testing HLA-B*5801 for patients of Southeast Asian and African descent). It is also conditionally contraindicated in patients with a history of a single gout flare or aHU (>6.8 mg/dL). Such patients should undergo a through comorbidity screening, especially toward hypertension, hyperlipidemia, and diabetes. For those requiring hypotensive treatment, loluoan, an angiotensin II receptor blocker, is an advantageous drug as it lowers UA by 20%–25% (Choi et al. 2012). A second important group of drugs with UA reducing effect are flozins – most studies show UA reduction by approximately 0.6 mg/dL (Sánchez-Briales et al. 2024). A modest uricosuric effect is also exerted by calcium channel blockers and fenofibrate.
There is no doubt that with the progression of CKD, sUA levels rise. However, evidence that HU exacerbates preexisting CKD is insufficient. The Uric Acid Right for Heart Health Study (cohort of 21,963 patients who were followed for 9.8 years) showed that patients with albuminuria generally have higher sUA levels (with an exception for macroalbuminuria and eGFR <45 mL/min), and that high sUA levels are a risk factor for cardiovascular and all-cause mortality independently and additively to the reduced GFR. Interestingly, the predictive power of sUA decreases along with the severity of renal impairment (Maloberti et al. 2023). The authors of the URRAH study conclude that HU most likely exacerbates kidney damage; however, this study was not designed to prove this particular cause–effect relationship.
In studies that include biopsy results, HU is considered an independent risk factor for moderate tubular atrophy/interstitial fibrosis and arteriolar damage in patients with IgA or membranous nephropathy (Russo et al. 2020; Liu et al. 2021b). Some histopathological features such as glomerulosclerosis, mesangial matrix expansion, endocapillary proliferation, interstitial fibrosis, and tubular atrophy were more common in patients with HU. However, multivariable linear regression has shown sUA significant correlation with those features only in female patients with immunoglobulin A nephropathy (IgAN) (Choi et al. 2021).
The heritability of the serum UA level is estimated at 30%–60%. A Nature paper by Tin et al. (2019) mentioned 183 susceptibility loci influencing UA traits. They can have either both hazardous or protective effects, and they vary with the HU type (ROL vs. RUE). For example, urate transporter gene 1 (URAT1/SLC22A12), a urate transporter gene, is a causative gene for renal hypouricemia type 1. In a study including 1993 patients with gout and 4902 healthy individuals, none of these variants were observed in the gout group. What is more, the URAT1 nonfunctional variants rs121907896 and rs121907892 resulted in a decrease of 2.19 mg/dL or 5.42 mg/dL in sUA in men and of 1.08 mg/dL or 3.89 mg/dL of sUA, respectively (Sakiyama et al. 2016). On the contrary, alleles rs3825018, rs7932775, rs475688, and rs3825016 of SLC22A12 are significantly associated with HU (Zheng et al. 2022). In ABCG2 gene, encoding the efflux pump BCRP, the rs2231142 allele has significant associations with both gout and HU (Chen et al. 2018). Interestingly, a missense allele (rs2231142) was linked to a reduced response to ALL (Wen et al. 2015). Not only the presence of such variants but also concomitant diseases matter. Certain SLC2A9 and ABCG2 variants, associated with urate levels, have a significantly greater effect on patients with CKD than on the general population (Jing et al. 2018). As most individuals with HU never develop gout, Kawamura et al. (2019) performed a comparative GWAS of HU risk loci and gout risk loci. They have proposed a two-step model of gout development: normouricemia to asymptomatic HU and asymptomatic HU to gout. Importantly, different loci are involved in these steps, suggesting that not only HU genes but also “HU to gout” genes are required for a person to develop gouty arthritis. Furthermore, this finding is supported by a metabolomic study, which shows differences in the profiles of individuals with HU and gout (Shen et al. 2021b). This partially explains why some patients with normal sUA levels present with UA-mediated inflammation (Kawamura et al. 2019). The three identified “HU to gout” paths included susceptibility loci: CNTN5, MIR302F, and ZNF724. These findings may be different in the European population as the cohort in Kawamura et al. (2019) study included only Japanese men. Interestingly, CNTN5 polymorphism is associated with anti-TNF response in Crohn’s disease (Thomas et al. 2014). In a different paper, the sUA level was significantly increased in patients with inflammatory bowel disease, and successful treatment of an active form of colitis resulted in a decrease of SUA (Zhu et al. 2019).
The -omics studies are also a promising filed for future research. So far, analysis of a metabolomic profile with mass spectrometry provides distinguishing features in patients with gout vs. aHU as they differ in metabolites involved in oxidation of fatty acids, carnitine synthesis, or urea cycle (Liu et al. 2023). What is more, the serum proteome seems to be different in patients with aHU, gout, and gout with kidney damage. Correlations show a link between abnormal plasma and immune-inflammatory response proteins (Shen et al. 2021a).
Approximately one-third of urate is excreted through the intestine. This active process involves an ABCG2 transporter, expressed in the intestinal epithelium and on the luminal surface of renal proximal tubule epithelial cells (Huls et al. 2008). Intestinal bacteria degrade urate almost completely. The influence of gout microbiota on sUA has only been tested on animal models, but studies show a decrease in sUA and lesser kidney injury in rats treated with probiotics (García-Arroyo et al. 2018; Xiao et al. 2020). Whether this has any significance for humans remains unknown.
Tamm–Horsfall protein also known as UMOD was first purified in 1952. It is a glycoprotein produced exclusively in the thick ascending limb of the loop of Henle by renal tubular cells. Its physiological function is only partially explained. However, it plays a crucial role in sodium balance and countercurrent gradient formation, prevents adhesion of Escherichia coli to the uroepithelium (Pak et al. 2001), and serves as a part of the immune system by binding immunoglobulin G light chains, complement components C1 and C1q, IL-1β, IL-6, IL-8, TNF-α, and interferon-γ (Wu et al. 2018). UMOD mutations cause familial juvenile hyperuricemic nephropathy, congenital urolithiasis, hereditary HU, and gout. UMOD deficiency in mice results in hypertension and HU (Liu et al. 2018a). Elevated UMOD concentrations, associated with a common polymorphism in the UMOD region, precede the onset of CKD (Köttgen et al. 2010). Serum UMOD was put forward as a biomarker of tubulointerstitial injury. As mentioned above, a proposed mechanisms in which UA could injure the kidneys include crystal deposition in the renal tubules, impairing tubular function and reducing UMOD secretion (Scherberich et al. 2018). A study on more than 100 Taiwanese patients with gout revealed that the spot urinary UMOD/creatinine ratio was significantly lower in patients with CKD than in those with normal kidney function. Whether this indicates a protective role of UMOD or just places it alongside other markers of CKD in patients with gout requires further investigation (Wu et al. 2019).
Elevated UA and resulting MSU crystal formation can trigger immune responses, leading to the activation of the NLRP3 inflammasome in macrophages, which in turn, induces the production of IL-23. As a pro-inflammatory cytokine, IL-23 increases the production of IL-17, contributing to the recruitment of neutrophils. IL-23 levels are heightened in patients with gout, but it is unclear whether the same could be said about patients with aHU. Activation of the NLRP3 inflammasome is suppressed by IL-37, reducing the release of IL-1β, inhibiting the activity of monocytes and macrophages. Serum IL-37 could be considered an inflammation marker as it is higher in active gout than inactive gout patients, with no significant difference between HU and normal controls (Ding et al. 2021). Administration of recombinant human IL-37 into murine models of gout downregulates pro-inflammatory cytokines such as IL-1β and IL-6, which may have potential for treatment (Liu et al. 2016).
The immunology of gout and HU gives much room for further considerations. For example, if neutropenic animals have a suppressed inflammatory response to MSU crystal injection, which is reversed after neutrophil repletion (Merriman 2024), do neutropenic patients need to worry about gout? As the expression of ABCG2 (urate transporter) or IL-1B genes is increased in patients with gout, it would be interesting to see whether this is also true in aHU (Natsuko et al. 2022). Another interesting issue is the influence of UA on AKI, as in vitro studies show that the concentration >10 mg/dL decrease the number of reactive oxygen particles and IL-6 production in macrophages (Gnemmi et al. 2022).
There are several groups of ULDs: xanthine oxidate inhibitors (XOI), which block a key enzyme in the UA production line, uricosuric agents, which block UA reabsorption in proximal renal tubules (probenecid and lesinurad), and drugs replacing uricase (pegloticase and rasburicase), helping to convert UA to soluble allantoin. All ULDs can cause gastrointestinal symptoms, headaches, and skin hypersensitivity reactions. XOI, mainly ALL and febuxostat (FBX), are considered the safest and best tolerated group. Mild rashes with ALL use are the main complaint; however, it can cause severe skin reactions such as Stevens–Johnson Syndrome or toxic epidermal necrolysis. ALL hypersensitivity syndrome is rare but potentially fatal, characterized by fever, eosinophilia, renal dysfunction, and rash. The dose has to be reduced in CKD, preventing many patients from achieving the target level of sUA. Although the official guidelines recommend an adjustment of ALL dose to eGFR, the maximum supportive dose in varying stages of CKD is not known. In patients with advanced CKD not achieving the UA goal, the addition of FBX, which has a higher gastrointestinal clearance, may be considered.
Probenecid can lead to the formation of UA stones as a consequence of increased UA excretion; it is also contraindicated in patients using salicylates or with blood dyscrasias due to the risk of aplastic or hemolytic anemia. Lesinurad can exacerbate CKD and cause acute renal failure (especially with inadequate hydration) and is contraindicated if eGFR is <30 mL/min or in advanced heart failure. Pegloticase and rasburicase infusions can be associated with shortness of breath, flushing, and chest discomfort and require premedication with antihistamines or corticosteroids. Severe allergic reactions may occur, particularly in patients with high anti-pegloticase antibody titers. The dose is independent of kidney function.
For many years, ALL was the drug of choice in the treatment of HU because of the higher all-cause mortality of patients treated with febuxostat (FBX) reported in one publication (White et al. 2018). However, this study had a serious downside of no placebo group, and the cardiovascular safety of FBX has since been revised (Pawar et al. 2021). FBX seems to be more effective than ALL in reducing sUA (Peng et al. 2020; Yang 2020), and in several studies, including a meta-analysis, patients on FBX have higher GFR than those on placebo after 6 months of treatment. Similarly, in a post hoc attribute-based analysis of the FEATHER study, FBX alleviated the decline in kidney function among stage 3 CKD patients with aHU without proteinuria (Kataoka et al. 2022). Combined with a novel drug, verinurad, it also reduces albuminuria in patients with type 2 diabetes (Stack et al. 2021).
Two large meta-analyses, one from 2021 of 9 randomized clinical trials (RCTs) and a second one from 2024 of 16 RCTs, shift toward confirmation of the nephroprotective effects of FBX. In the first one, the FBX treated group showed a higher eGFR at 6 months with a weighted mean difference (WMD) of 2.86 mL/min/1.73 m2 (p < 0.001) (Chewcharat et al. 2021). The second study also showed a slower decline in eGFR, though with a WMD of 0.90 mL/min/1.73 m2, with most studies following patients for at least 6 months. FBX use also reduced the urine albumin to creatinine ratio and was associated with a reduced risk of kidney events (Yang et al. 2024). Similar slowing of eGFR decline can be observed with uricosuric treatment with benzbromarone (Kohagura et al. 2023). Importantly, both quoted meta-analyses lack the division between patients with gout and aHU (with an exception for reduced risk of kidney events, which was confirmed in both HU and gout groups). FBX has good renal safety outcomes in almost all analyzed studies (Baek et al. 2018; Kim et al. 2020; Liang et al. 2022) and seems to be more effective than ALL in preserving renal function in CKD stage 5 patients (Hsu et al. 2020).
Though effective in lowering sUA and preventing gout flares, new drugs, such as anakinra or canakinumab, need further safety studies in patients with CKD, especially in grade 3b and lower (Pisaniello et al. 2021). New guidelines will probably include sodium-glucose cotransporter-2 inhibitors as their effect on lowering UA was reported in 43 randomized controlled trials (Yip et al. 2022). As this group has recently conquered the field of heart insufficiency therapy, it will be interesting to see whether the cardioprotective effect is in any way mediated by lowering UA concentrations.
The question whether HU treatment confers a nephroprotective effects remains open. Randomized controlled trials and meta-analysis of thereof bring conflicting results (Table 1). A meta-analysis published in 2020 did not show benefits on the incidence of kidney failure but did report a 0.68 mL/min/1.73 m2/year difference in eGFR slope for trials with a follow-up of at least 2 years (Chen et al. 2020). However, as mentioned by Gonzalez-Martin et al. (2020), included studies were not limited to patients with CKD, and no comparison with age-associated loss of eGFR was available. He also noticed that the futility analysis in a large trial considered a clinically meaningful difference to be 0.6 mL/min/1.73 m2/year, which would make the result marginally higher. Other comparative studies also fail to notice a significant improvement in kidney function (Liang et al. 2022; Wu et al. 2022). On the contrary, in a recent meta-analysis by Luo et al. (2024) comprising 17 studies published between 2006 and 2023, it was concluded that between ULT group and placebo/no treatment, in both long-term (>6 months) and short-term (3–6 months) groups, ULT conferred a nephroprotective effect. In the long-term group, patients receiving ULT had a 2.07 mL/min/1.73m2 lesser decline in eGFR compared to those receiving a placebo or no treatment (Luo et al. 2024).
Comparison of meta-analyses published between 2022 and 2018 with various treatment regimens for HU in CKD against each other or placebo. Trials including patients with gout were not mentioned
| Year | UA (mg/dL)a | CKD stage | No. of ptsb | Drugs | End pointsc | Conclusion | Follow-up, months | Reference |
|---|---|---|---|---|---|---|---|---|
| 2024 | 6.3 ± 1.49 | G2–G4 | 2516 | FBX | eGFR | Favoring FBX | 3–12 | Yang et al. (2024) |
| 2023 | >6 | G2–G5 | 1521 | ALL/FBX | ΔDGFR | Favoring ULT | 3–36 | Liu et al. (2018b) |
| 2022 | 7 | G1–G5 | 447 | ALL/FBX/BZB | eGFR | Favoring ALL | >6 | Tien et al. (2022) |
| 2022 | NS | G1–G4 | 722 | ALL |
|
| 21–38 | Wu et al. (2022) |
| 2022 | NS | G2–G5 | 1249 | ALL/FBX/TPX | eGFR | No impact | 5–84 | Yu et al. (2022) |
| 2022 | Variousd | Various | 866 | ALL | 24HP | Decreased | 2–36 | Luo et al. (2022) |
| 2022 | 5.96 ± 1.21 | G2–G4 | 1469 | ALL/FBX | eGFR | No impact | 4–27 | Liang et al. (2022) |
| 2022 | 7.26 ± 0.15 | G1–G4 | 3095 | ALL/FBX |
|
| 4–36 | Zhang et al. (2022) |
| 2022 | 7/NA | G2–G4 | 1480 | TPX/FBX/ALL |
|
| 3–24 | Tsukamoto et al. (2022) |
| 2022 | Various | Various | 3209 |
|
|
| 0.5–84 | Sapankaew et al. (2022) |
| 2021 | 4.61 (F)/5.5 (M)/NI | G2–G4 or NI | 1943 | ALL/FBX/BZB |
|
| 3–27 | Liu et al. (2021a) |
| 2021 | 7 | Various | 5439 | ALL/FBX/TPX/BZB |
| Favoring ULT | >12 | Sharma et al. (2021) |
| 2021 | 5.5 | Various | 2141 | FBX | eGFR | Increased | 6–NI | Chewcharat et al. (2021) |
| 2019 | 4.61 | G2–HD | 1317 | FBX | eGFR | Increased | 1–25 | Lin et al. (2019) |
| 2019 | NS | Various | 402 | FBX | eGFR | Increased | 1–12 | Lin et al. (2019) |
| 2018 | 5.46 ± 1.25 | G2–G4 | 437 | FBX | eGFR | Increased | 3–48 | Zeng et al. (2018) |
| 2018 | NI | G1–G4 | 832 | ALL/FBX | eGFR | Favoring ULT | 4–24 | Liu et al. (2018b) |
Lowest UA included in a single study.
Includes placebo group.
Selected end points.
Various – meta-analysis includes people of all stages of CKD and healthy subjects.
FBX improved eGFR in CKD.
ALL, allopurinol; BZB, benzbromarone; CKD, chronic kidney disease; CRE, composite renal events: deterioration of renal function, end-stage renal disease, and initiation of renal replacement therapy; eGFR, estimated glomerular filtration rate; FBX, febuxostat; GFR, glomerular filtration rate; NI, no information; NS, no significance difference; pts, patients; TPX, topiroxostat; UA, uric acid; UAER, urinary albumin excretion rate; ULT, uric acid lowering therapy; 24HP, 24-hour proteinuria.
Such differences may be explained by several factors. First, in their attempt to increase the power of a study and include as many papers as possible, the participant heterogeneity is significant: meta-analyses include studies combining patients with aHU and gout or with CKD vs. normal renal function and various sUA levels. Most studies do not include genomic data, so no differentiation between those groups with high-risk alleles and those without is possible. Second, the follow-up time is usually no longer than 2–3 years. As noted by Gonzalez-Martin et al. (2020), the difference in eGFR between the urate-lowering and control groups decreases over time in other studies. Patients treated with ULT often stay on it for at least 10 years, which needs to be weighed against potential side effects and interactions (for example, with amoxycillin, vitamin K antagonists, cyclosporin, and ACE-I). During yearlong use, such interactions are frequently overlooked. Third, changes in creatinine and eGFR might not be sufficient data to draw conclusions on the nephroprotective effect. Just as major adverse cardiovascular events are more important than pro-B-natriuretic peptide levels in cardiology, strong end points such as end-stage kidney failure, dialysis, and mortality should be the focus of future studies.
Summing up, the nephroprotective effect of ULT is probably mild and needs to be balanced against safety concerns regarding various drug types.
Despite the abundance of research papers on the subject, the views on aHU vary and it seems like every team approaches the issue using different statistical methods. With thousands of published studies and no clear conclusion, we need to ask ourselves: do we need more data? or do we just need better data? Currently, there is no evidence that aHU leads to CKD; rather these two are frequently met comorbidities. With safety concerns regarding UA lowering agents in causing AKI with no clear benefits on eGFR in the long perspective, further use might result in more cons than pros. In our opinion, future studies on the topic should not only include sUA level but also consider renal parameters such as FEUA and measured GFR. Genomic analysis should become standard in HU research; it would be also of value to design randomized controlled trials, evaluating the impact of ULT in genetically defined subgroups, both in patients with CKD and without. Additionally, including -omic technologies in stratifying patients based on their risk of progression could be a step forward in implementing personalized medicine in ULT. As mentioned earlier, significant differences exist between patients with aHU and gout in context of metabolomics; exploring those differences could help identifying new therapeutic targets and response to treatment. Instead of classical statistics and metanalysis, we could try to employ machine learning in data analysis. The goal is to define certain groups of patients who will benefit from urate-lowering therapy, not only by decreasing the downward slope of eGFR but also in strong end points such as reduced mortality and dialysis incidence.