
Fig. 1
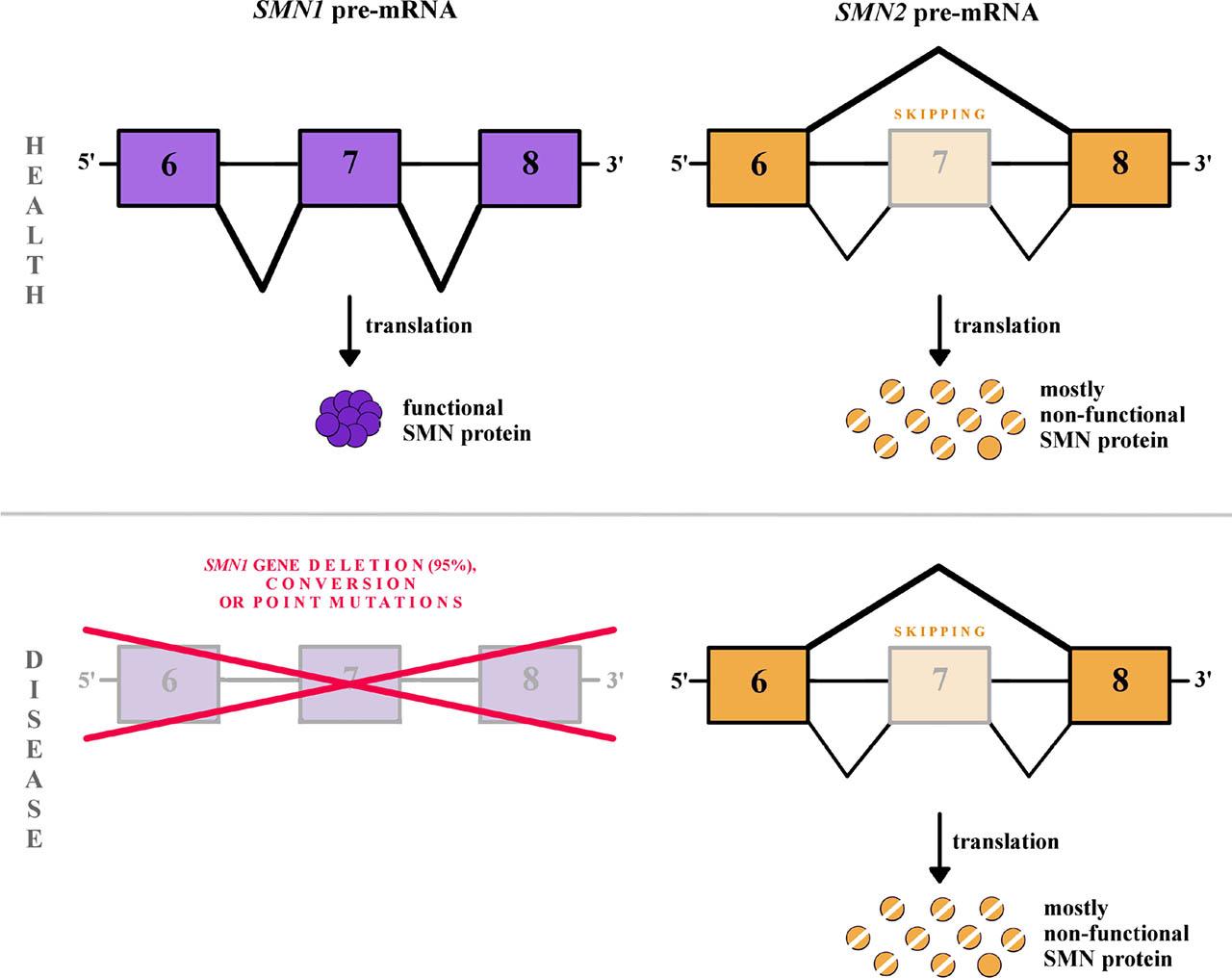
Fig. 2
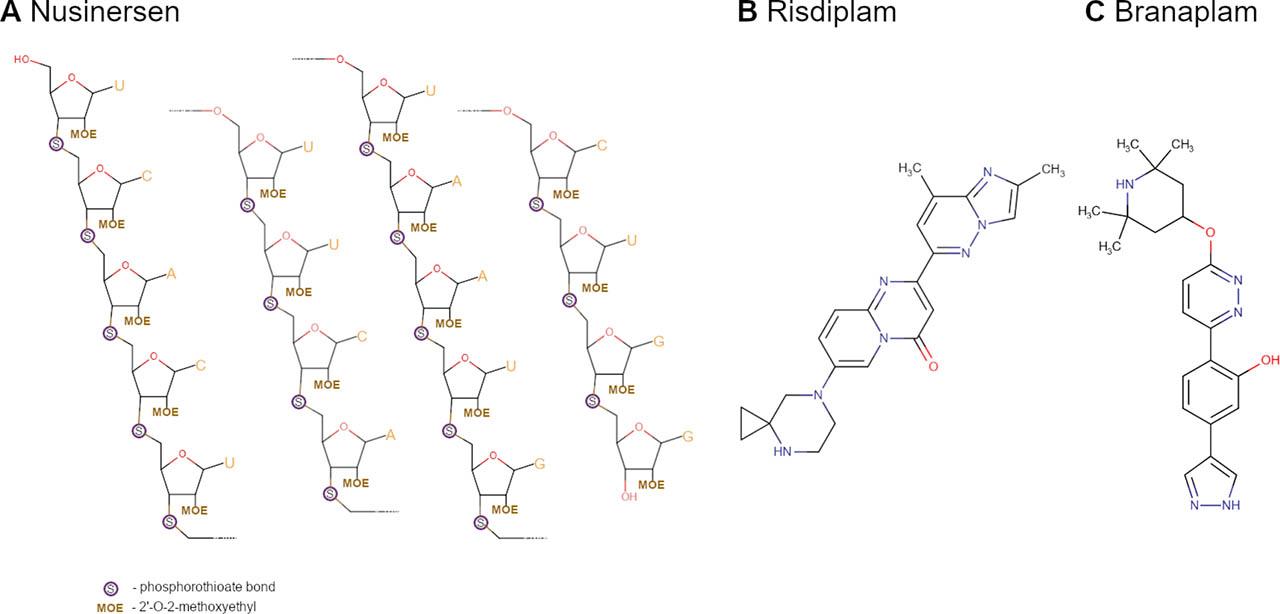
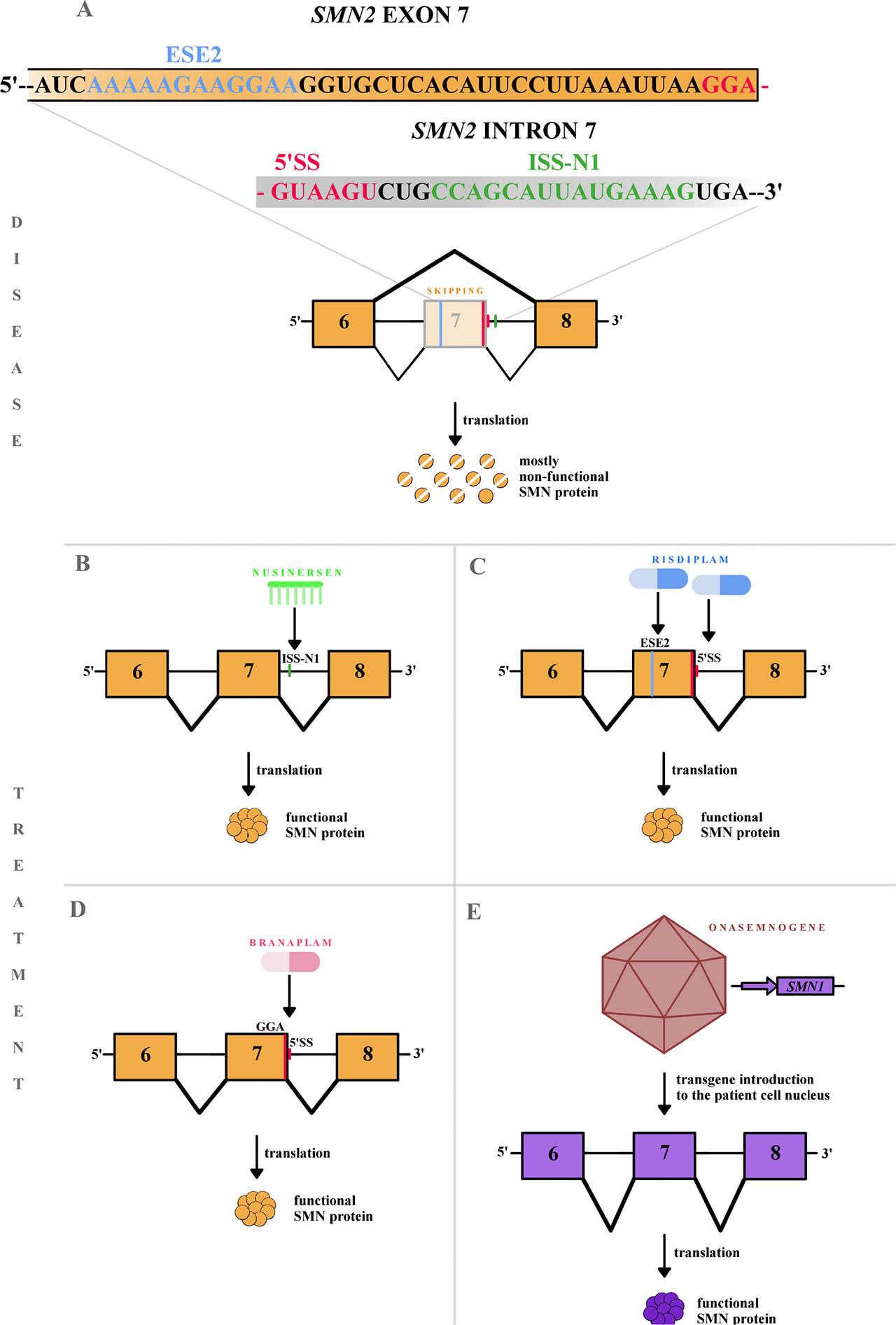
Fig. 3
Summary of risdiplam clinical trials Roche_ Roche announces 2-year risdiplam data from SUNFISH and new data from JEWELFISH in infants, children and adults with spinal muscular atrophy (SMA) [29, 30, 31, 32, 33] SMN1- survival of motor neuron; SMN2- survival of motor neuron 2; MFM- Motor Function Measure
| Clinical trial | Identification number | Characteristics | Patients profile | Main purpose of study | Current status of study |
|---|---|---|---|---|---|
| FIREFISH | NCT02913482 | Two-part, open label study. | Infants aged 1–7 months of age with SMA type I and two SMN2 gene copies. | Part one: dose-escalation study | The study met its primary endpoint. |
| SUNFISH | NCT02908685 | Two-part, double blind, placebo-controlled study. | People between 2–25 years old with SMA type II or III. | Part one: dose-escalation study | The study met its primary endpoint. |
| JEWELFISH | NCT03032172 | Open-label exploratory trial. | People between 6 months – 60 years, previously treated with SMA-directed therapies | Safety and tolerability of daily risdiplam dose in non-naïve patients who have taken nusinersen, olesoxime or onasemnogene abeparvovec-xioi. | The study has completed recruitment. |
| RAINBOWFISH | NCT03779334 | Single-arm, multicentre study. | Babies from birth to six weeks of age (at first dose) with genetically diagnosed SMA, without symptoms. | Efficacy, safety, pharmacokinetics and pharmacodynamics. | The study is currently in phase 2. No results were published yet. |
Summary of approved therapeutics and substances under clinical trials for SMA [20, 23, 24, 28, 35, 36, 38]
| Therapeutic | Management company | Characteristics | Patient's profile | Dosage and administration |
|---|---|---|---|---|
| Spinraza** | Biogen | Survival motor neuron 2 (SMN2) splicing modifier | All ages and types | Administered by intrathecal injection at an equivalent dose of 12 mg (4–5 ml based on age) |
| Zolgensma** | Novartis Gene Therapies* | Gene therapy based on using recombinant adeno-associated virus subtype 9 (AAV9) to overexpression SMN1 gene | Less than two years old paediatric patients with spinal muscular atrophy and bi-allelic mutations in the SMN1 gene | One intravenous infusion; dosage based on patient's body weight |
| Evrysdi** | Roche, Genetech Inc. | Survival motor neuron 2 (SMN2) splicing modifier | Patients 2 months of age and older suffering from SMA | Administrated orally once a day; dosage is determined by patient age and body weight |
| Reldesemtiv | Cytokinetics/Astellas | Muscle drug (non-SMN) | Patients with SMA types 2, 3 and 4 who are age 12 or older | Administrated orally; phase 2 trial completed |
| Apitegromab | Scholar Rock | Muscle drug (non-SMN) | Patient between 2 and 21 who have SMA type 2 or 3. | Administrated by intravenous infusion; phase 2 trial ongoing |